Article Text

Abstract
Background Pulmonary hypertension (PH) causes increased morbidity and mortality in patients with interstitial lung diseases (ILD). Classification schemes, while well-characterised for the vasculopathy of idiopathic PH, have been applied, unchallenged, to ILD-related PH. We evaluated pulmonary arterial histopathology in explanted human lung tissue from patients who were transplanted for advanced fibrotic ILD.
Methods Lung explants from 38 adult patients who underwent lung transplantation were included. Patients were divided into three groups: none, mild/moderate and severe PH by mean pulmonary artery pressure (mPAP) measured at pre lung transplantation right heart catheterisation (RHC). Grading of pulmonary vasculopathy according to Heath and Edwards scheme, and prelung transplantation evaluation data were compared between the groups.
Results 38 patients with fibrotic ILDs were included, the majority (21) with idiopathic pulmonary fibrosis. Of the 38 patients, 18 had severe PH, 13 had mild/moderate PH and 7 had no PH by RHC. 16 of 38 patients had severe pulmonary arterial vasculopathy including vascular occlusion with intimal fibrosis and/or plexiform lesions. There were no correlations between mPAP and lung diffusion with the severity of pulmonary arterial pathological grade (Spearman’s rho=0.14, p=0.34, rho=0.11, p=0.49, respectively).
Conclusions Patients with end stage ILD had severe pulmonary arterial vasculopathy in their explanted lungs irrespective of the presence and/or severity of PH as measured by RHC. These findings suggest that advanced pulmonary arterial vasculopathy is common in patients with advanced fibrotic ILD and may develop prior to the clinical detection of PH by RHC.
- interstitial fibrosis
- primary pulmonary hypertension
- lung transplantation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
What is the pulmonary arterial vascular histopathology in patients with end stage fibrotic interstitial lung disease and does it correlate to measured pulmonary arterial pressure?
What is the bottom line?
One-fifth of patients with end stage fibrotic interstitial lung disease had severe pulmonary arterial vasculopathy including plexiform lesions in their explanted lungs.
Why read on?
This study provides direct evidence that WHO group 3 PH related to hypoxemic lung disease has histopathological similarities to WHO group 1 pulmonary arterial hypertension. These findings suggest that severe pulmonary arterial vasculopathy is more common than previously recognised and may develop in advanced fibrotic interstitial lung disease prior to the clinical detection of pulmonary hypertension.
Introduction
Pulmonary hypertension (PH) is a well-known cause of increased morbidity and mortality in patients with interstitial lung diseases (ILD).1–3 ILD-related PH, a subset of the WHO group 3 PH, is defined by a mean pulmonary artery pressure (mPAP) greater than 25 mm Hg on right heart catheterisation (RHC). The prevalence of PH in fibrotic ILD increases with the severity of the lung disease. PH occurs in 46% of end stage fibrotic lung disease awaiting lung transplantation and severe PH, defined as mPAP >35–40 mm Hg is observed in 5%–10%.2–9
The modern clinical classification of PH is five-group taxonomy based on the attributable cause and anticipated vascular histopathology. Group 1 is pulmonary arterial hypertension (PAH) (including idiopathic PAH (iPAH)) while groups 2–5 are aetiologically different and are thought not to derive from primary arterial vasculopathy. Plexiform vasculopathy is the defining pathological feature of iPAH.10 11 Abundant human necropsy tissue studies have demonstrated plexiform arterial lesions since iPAH was first described in 1891 by Ernst Von-Romberg.12–14 Therefore, it is generally assumed that the plexiform lesion is a marker of advance WHO group 1 PAH and a correlate of elevated pulmonary vascular resistance (PVR). The classification of PH related to lung disease outside of primary vasculopathy comes from an assumed causal role of hypoxemia as opposed to a plexiform vasculopathy.15 Human tissue studies from well characterised group 3 patients are rare and these historical assumptions and modern classification scheme have stood unchallenged.16 17
We sought to evaluate the pulmonary arterial histopathology in explanted human lung tissue from well characterised patients who were transplanted for advanced fibrotic ILD. We aimed to examine patient clinical characteristics including pulmonary haemodynamics and their relation to pulmonary arterial histopathology including the presence or absence of plexiform arterial vasculopathy.
Materials and methods
Study population
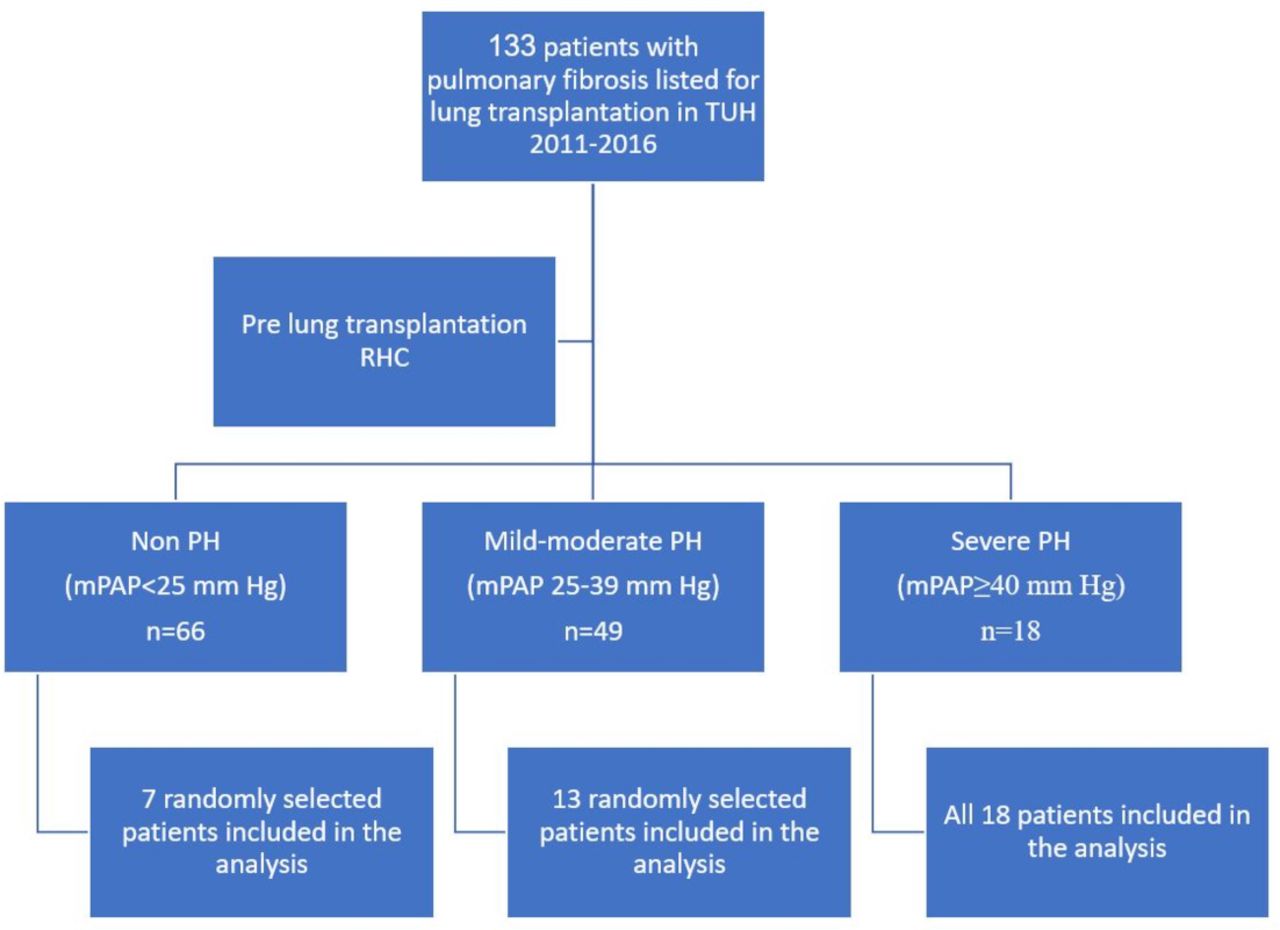
Data were obtained from 38 of the 133 (age≥18 years) patients with fibrotic ILD who received lung transplantation at Temple University Hospital (TUH), Philadelphia, USA, from 2011 to 2016. The patients were divided into three groups of measured mPAPs determined by pretransplant RHC (see below): none (n=66), mild-moderate (n=49) and severe PH (n=18). We included in the analysis all 18 patients with severe PH, 13 randomly selected patients with mild-moderate PH and 7 randomly selected patients without PH whose explanted lungs were available for pathological studies (figure 1). Clinical data were available as part of routine prelung transplantation evaluation included demographics, pulmonary function tests (PFTs), 6 min walk test, echocardiography, RHC and high-resolution CT.

Study design. mPAP, mean pulmonary artery pressure; PH, pulmonary hypertension; RHC, right heart catheterisation; TUH, Temple University Hospital.
Grading of PH severity
PH was classified by pretransplantation RHC values—mPAP and pulmonary arterial wedge pressure (PAWP) according to the recent haemodynamic definitions and updated clinical classification of PH guidelines by the 6th World Symposium on Pulmonary Hypertension Task Force: Precapillary PH defined by mPAP>20 mm Hg and PVR ≥3 Wood units (WU) and PAWP≤15 mm Hg.18 PH severity was graded by mPAP: non-PH—patients with mPAP ≤20 mm Hg; mild/moderate PH—patients with 20<mPAP<40 mm Hg, PVR ≥3 WU and PAWP≤15 mm Hg and severe PH—patients with mPAP ≥40 mm Hg, PVR ≥3 WU and PAWP≤15 mm Hg.18 PVR was available (WU) but was not used for grading of PH severity.
Tissue acquisition procedure
All fixed and frozen tissues were obtained as part of the Lung Tissue Research Consortium (LTRC). The LTRC is sponsored by the American National Heart Lung and Blood Institute and TUH is one of the five clinical centres for LTRC in the USA. The tissues were preserved within 30 min of removal from the patients. Briefly, tissues were placed in liquid N2 and once completely frozen transferred to a container filled with dry ice or a −80°C freezer. Then, it was covered with RNAlater and totally immersed in fixative with 10% neutral buffered formalin. Slides were made with H&E and pentachrome staining from the formalin-fixed specimens from each lobe. All tissue specimens were sent to the Tissue Core Laboratory and stored as follows: RNAlater tissue was aliquoted and stored at −20°C and Flash (snap) frozen tissue was aliquoted and stored at −80°C.
Grading of hypertensive pulmonary vascular disease
Paraffin block specimens were prepared from lung tissue as described above. Sections were visually scored at 10× magnifications. Grading of the pulmonary vasculature was done blinded to the mPAP (author WH) according to Heath and Edwards’s scheme for hypertensive pulmonary vascular disease with progressive structural changes10 (table 1). All vessels seen on each slide were examined. However, vascular grading was applied only to pulmonary arterioles. Venules were examined to rule out signs of pulmonary veno-occlusive disease.
Heath and Edwards’s scheme for hypertensive pulmonary vascular disease with progressive structural changes10
Due to the high unexpected frequency of plexiform lesions, a second pathologist (author AA) reviewed all specimens, and only cases that were diagnosed by both pathologists as plexiform lesions were included in the study as such (8/11).
Statistical analysis
Data are presented as mean±SD for continuous variables or counts (percentages) for categorical variables. χ² was used for categorical data, and the paired or unpaired t-test was used for continuous data. The Spearman rank-order correlation coefficient was used to measure the strength and direction of association between continuous and ordinal variables. All data were analysed using SPSS V.12.0 (SPSS, Chicago, Illinois, USA) and Stata 14.0 (Stata, College Station, Texas, USA).
Patient and public involvement statement
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
This study was conducted in accordance with the amended Declaration of Helsinki. TUH review board approved the protocol, and written informed consent was obtained from all patients (protocol number 24556).
Results
Patients
A total of 38 patients with fibrotic end stage lung disease who had lung transplantation at TUH from 2011 to 2016 were included in the final analysis (figure 1). The average age of the patients was 65±8 years, 26 of them were males and 12 were females. The lung diseases of the patients were diverse, with 21 patients with idiopathic pulmonary fibrosis (IPF), 8 patients with combined pulmonary fibrosis and emphysema (CPFE), 3 patients with connective tissue disease related ILD, 2 patients with chronic hypersensitivity pneumonitis, 2 patients with interstitial pneumonia with autoimmune features, 1 patient with desquamative interstitial pneumonia) and 1 patient with pulmonary Langerhans cell histiocytosis. Their PFTs showed severe restrictive pattern with forced expiratory volume in the first second (FEV1) to forced vital capacity (FVC) ratio of 78%±14% and FVC 53%±19% of predicted, total lung capacity (TLC) 53+15% of predicted and lung diffusion for carbon monoxide (DLCO) of 22%±10% of predicted (table 2).
Patient characteristics grouped by PH severity
Pulmonary hypertension data
All patients had prelung transplant RHC. Patients were grouped by pretransplant PH severity derived from RHC measurement: 18 patients had severe PH with mPAP of 49±12 mm Hg, 13 patients had mild/moderate PH with mPAP of 26±2 mm Hg and 7 patients did not have PH (non-PH group) with mPAP of 17±3 mm Hg (p<0.0001 for all between groups comparisons). Age, gender and PFTs of all three groups were not statistically different (table 2). PVR was 9.1±6 WU for the severe PH group, 3.4±1.1 WU for the mild/moderate PH group and 2.8±1.4 WU for the non-PH group (p<0.01 for comparison of severe to mild-moderate and non-PH groups).
Vasculature pathology grade
All 38 patients had pulmonary artery histopathological changes ranging from grade 1 (distal extension of muscle into distal arterioles and medial thickening of muscular arteries) to grade 4 (progressive generalised arterial dilatation lesions, including plexiform lesions) (figure 2). 14 of 38 (37%) had grade 2 (medial hypertrophy with cellular intimal proliferation in small muscular arteries). Sixteen of 38 (42%) had grade 3 (progressive fibrous vascular occlusion, concentric intimal fibrosis) and/or grade 4 changes (figure 3). The mean vasculature pathology grade was 2.42±1.05 with no significant difference between all three groups of PH severity. There was no correlation between the presence and severity of PH (mPAP and PVR) and the severity of the vasculature pathology grade (Spearman’s rho=0.14, p=0.34 and Spearman’s rho=0.018, p=0.91, respectively, figure 4). There was also no correlation between the DLCO and the severity of the vasculature pathology grade (Spearman’s rho=0.11, p=0.49). Pulmonary venules in each slides were examined for signs of veno-occlusive disease which were not seen in any patient.
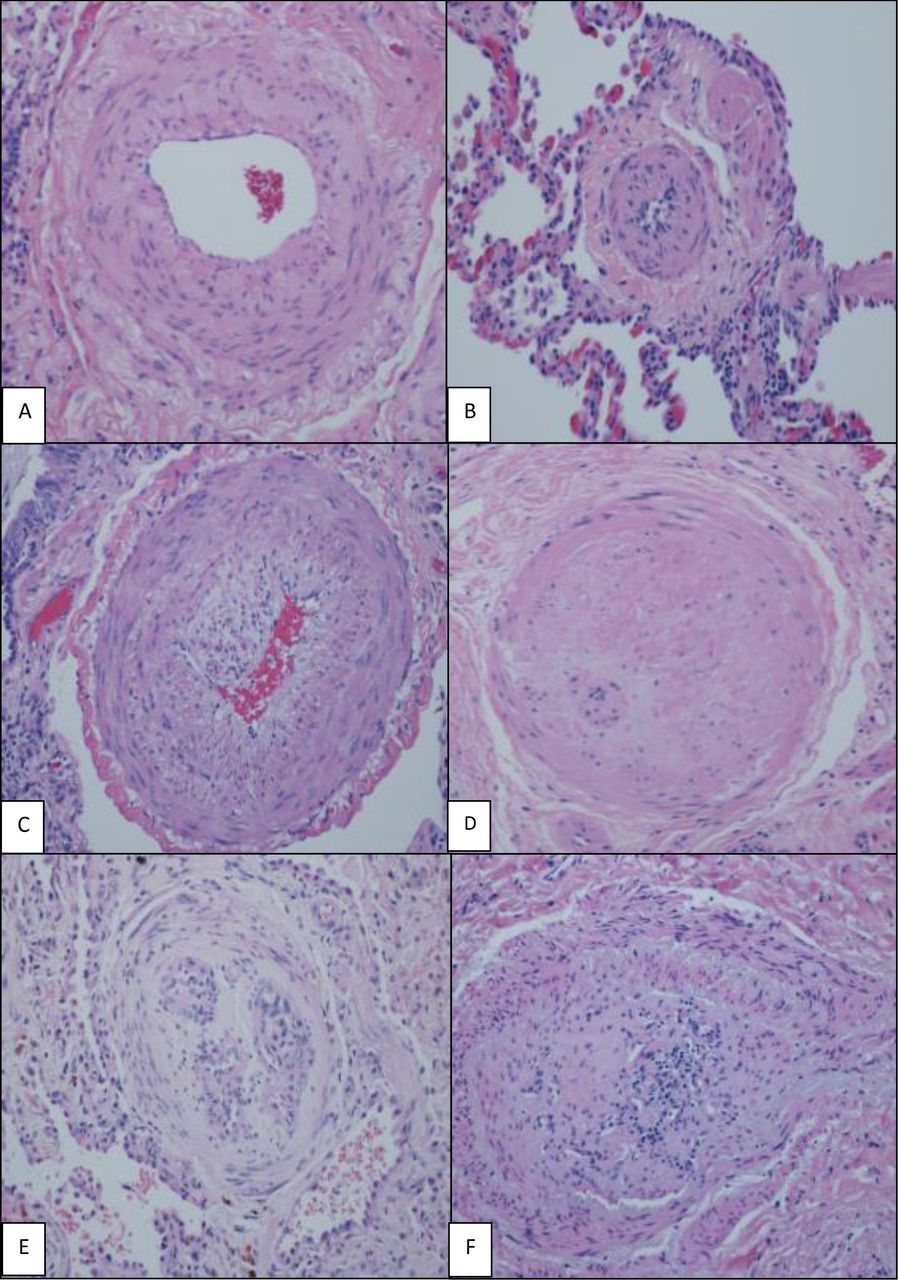
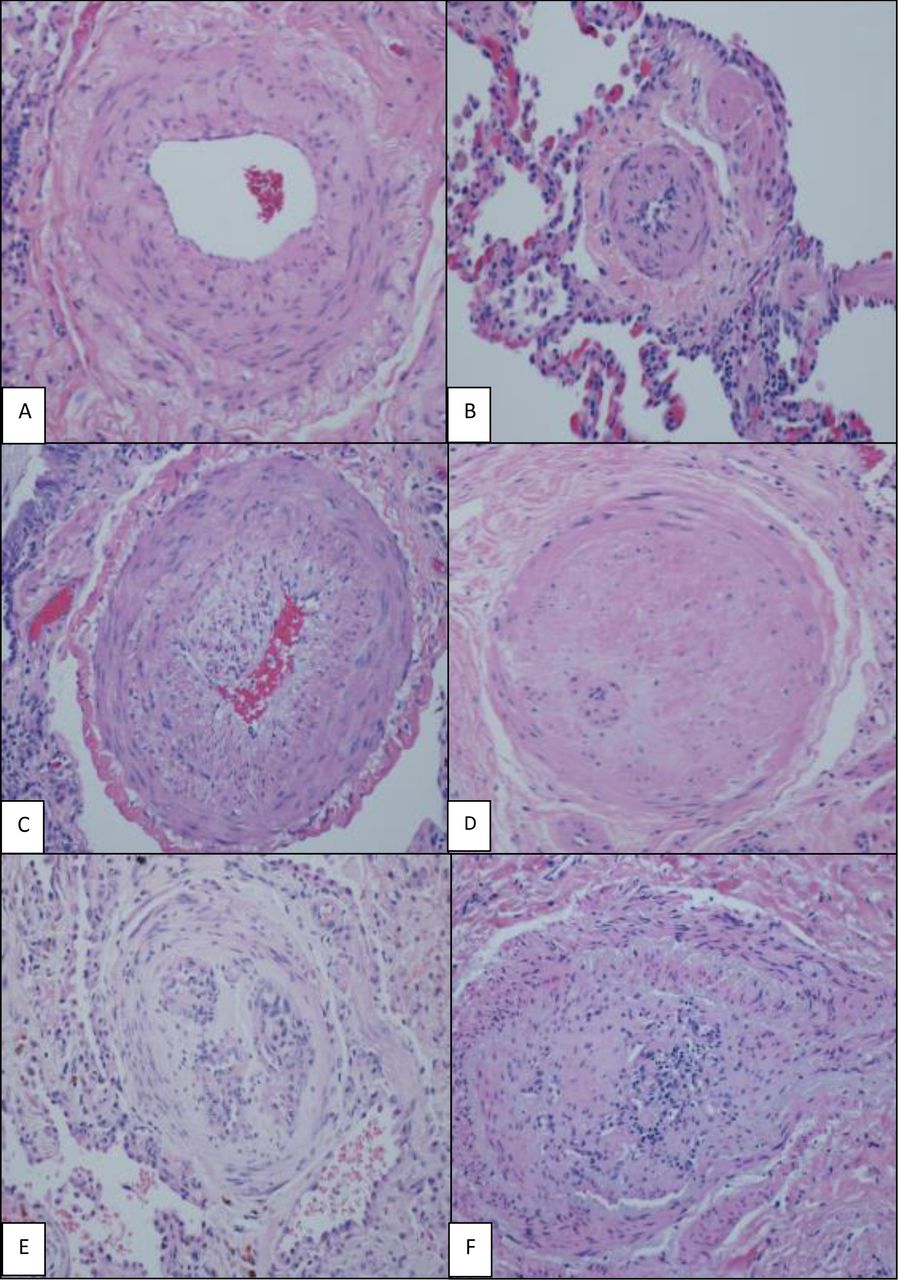
H&E (×200), various grades of histological changes (A–F). (A) Muscular artery with medial hypertrophy (increased number of smooth muscle nuclei arranged in parallel around the lumen). (B) Muscularisation of arterioles (thick muscle layer in arterial wall composed of numerous smooth muscle cells with closely spaced nuclei). (C) Intimal proliferation and fibrosis in addition to medial hypertrophy. (D) Concentric intimal fibrosis resulting in circumferential luminal narrowing. (E,F) Plexiform lesions; concentric intimal fibrosis causing near luminal obliteration and multiple small vascular spaces and expansion and partial destruction of the arterial wall with extension of the lesion into the perivascular connective tissue.
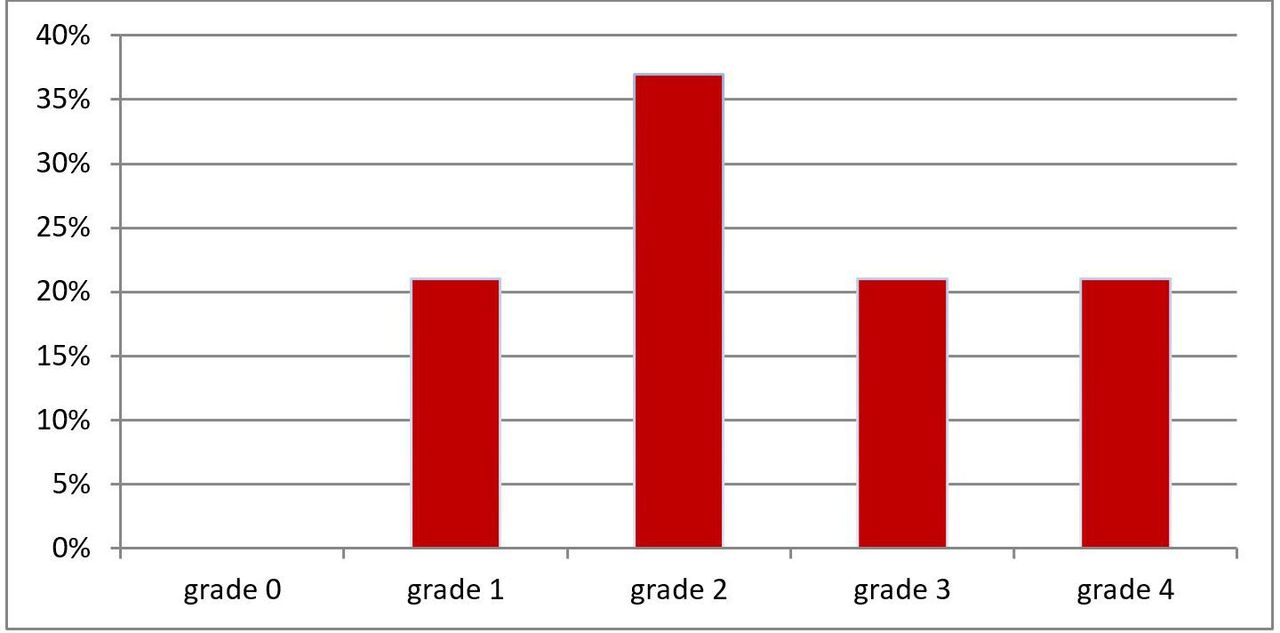
Pulmonary vasculopathy grades for all patients (n=38). Briefly, Grade 0—normal; Grade 1—medial thickening; Grade 2—medial hypertrophy; Grade 3—concentric intimal fibrosis; Grade 4—plexiform lesions (for full description, see Heath and Edwards scheme, table 1).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
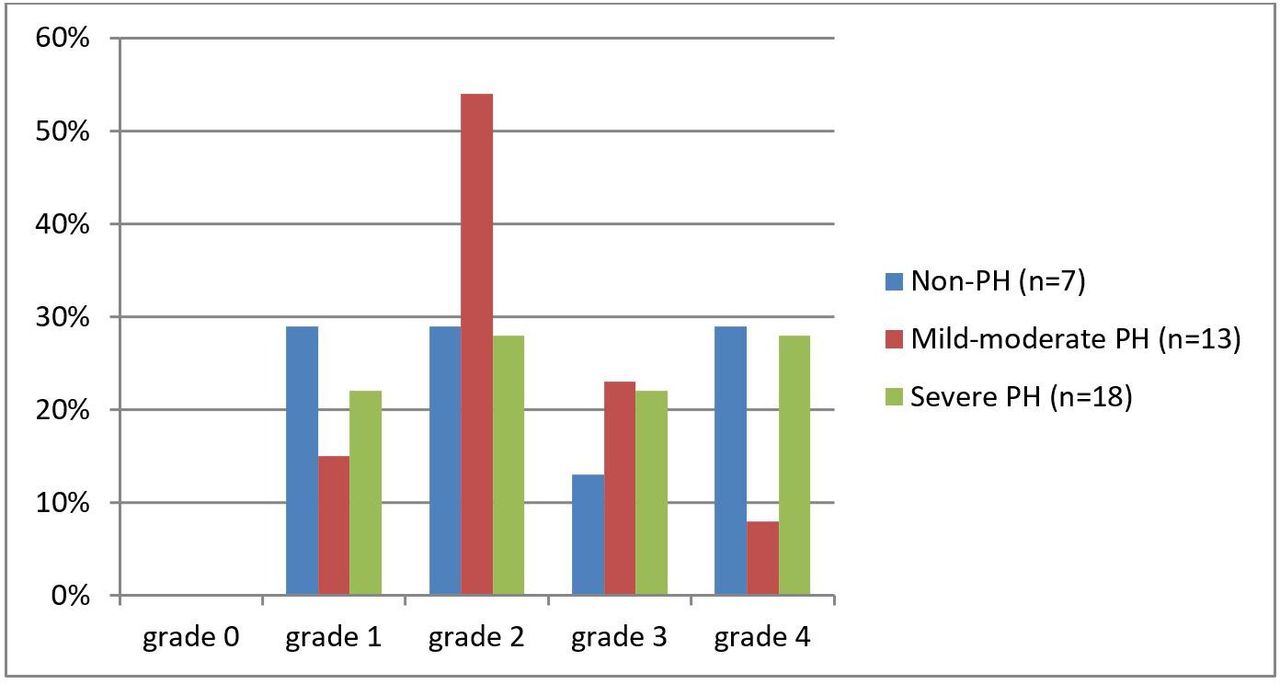
Pulmonary vasculopathy grades by PH severity (none, mild-moderate and severe PH). Briefly, Grade 0—normal; Grade 1—medial thickening; Grade 2—medial hypertrophy; Grade 3—concentric intimal fibrosis; Grade 4—plexiform lesions (for full description, see Heath and Edwards scheme, table 1). PH, pulmonary hypertension.
Plexiform lesions
Eight of 38 (21%) patients had plexiform pulmonary arterial lesions. Plexiform lesions were evident in all three PH severity groups: 2 of 7 non-PH, 1 of 13 mild/moderate PH and 5 of 18 severe PH. A comparison of patients with plexiform lesions (n=8, grade 4) to patients without plexiform lesions (n=30, grades 1–3) showed no statistically significant differences in pulmonary haemodynamics or clinical data including age, gender, PFTs and RHC data (table 3).
Comparison of patients’ characteristics between patients with and without plexiform lesions
Pulmonary vasodilators and antifibrotic therapy data
Eight of 38 (21%) patients were taking pulmonary vasodilator medication prior to lung transplant. Therapy included phosphodiesterase-5 (PDE-5) inhibitor (n=7), combination PDE-5 and endothelin receptor antagonist (n=4) and 1 prostanoid +dual therapy. As expected, mPAP was significantly higher in the group on baseline PH therapy (51.5±15.2 vs 30.7±13.2 mm Hg, p=0.002) as was PVR (9.89±6.46 vs 4.76±4.08 WU, p=0.04). Ten patients (26%) were taking antifibrotic therapy prior to lung transplant: pirfenidone (7/10) and nintedanib (3/10). Those taking antifibrotics had significantly lower mPAP and PVR (25.7±8.4 vs 39.3±16.7 mm Hg, p=0.001 and 4.22±2.5 vs 6.69±5.9 WU, p=0.04, respectively).
Discussion
Our study showed that patients with end stage fibrotic ILD undergoing lung transplantation had significant and often severe pulmonary arterial vasculopathy in their explanted lungs. Unexpectedly, this arterial vasculopathy was present in lungs from patients with and without PH and had no correlation to the clinical severity of the underlying lung disease. To the best of our knowledge, this is the first study to describe a high frequency of severe vascular changes in advanced fibrotic ILD, with almost half of the patients showing vascular occlusion with intimal fibrosis and one-fifth showing plexiform lesions which are traditionally considered a hallmark only of WHO group 1 iPAH pathology.
Our patients with PH are classified as WHO Group 3, PH secondary to hypoxemic lung disease. One of the most important factors contributing to pulmonary arterial changes in patients with ILD is chronic hypoxia, which is a part of the terminology of WHO group 3 (PH due to lung diseases and/or hypoxia). Chronic hypoxia affects all three layers of the pulmonary vasculature: the intima, media and adventitia and their harbouring cells—the endothelial cells (EC), smooth muscle cells (SMC) and fibroblasts, respectively.19 However, hypoxia alone does not usually lead to high-grade vascular remodelling with intimal obliteration and plexiform lesions as seen in iPAH.20
Plexiform lesions are networks of vascular micro vessels that consist of two distinct populations of EC: normal phenotype that lines vessels and the proliferating, apoptosis-resistant phenotype at the core of the lesion.21 22 They are considered the hallmark of severe iPAH and were first described in 1927.12 23,12 In 1989, a landmark study by Pietra et al, of 48 patients with iPAH documented plexiform lesions in 52% of the total cohort.12 This study also demonstrated that presence of plexiform lesions portended a poorer prognosis. In 2012, Stacher et al evaluated explanted lungs from 62 patients with group 1 PAH and documented plexiform lesions in 90% overall and in all of 48 patients with idiopathic group 1 iPAH.11
Data obtained by computerised three-dimensional reconstructions of serial lung sections from patients with PH, suggested that a single plexiform lesion can occlude the entire length of an affected vessel, contributing to PH severity.21
Historically, pulmonary arterial vascular changes are less well documented and infrequently described in patients with fibrotic ILD. A Japanese lung necropsy study of 60 patients with emphysema, IPF or CPFE reported plexiform arterial lesions in 2 patients with CPFE and 1 patient with IPF and none in emphysema.16 In a similar study of explanted lungs of 70 patients transplanted for end stage COPD (chronic obstructive pulmonary disease), only 1 patient had plexiform lesions.17 Our data demonstrated much more common advanced vasculopathy including plexiform lesions in end stage fibrotic ILD lung explants obtained at time of transplantation.
Our hypothesis was that patients with severe PH would have more severe pulmonary arterial vascular changes but not plexiform lesions seen in group 1 PAH. We were expecting positive correlation between mPAP and pulmonary artery vasculopathy grade and only mild to moderate vascular changes. However, pretransplantation measured mPAP and DLCO failed to predict which patients had pulmonary arterial pathological changes. In addition, we found unexpectedly severe vascular change and a high prevalence of plexiform arterial lesions in patients with normal directly measured pulmonary pressures. We found that severe pulmonary arterial vasculopathy was common in patients with end stage fibrotic ILD and that these changes could not be predicted clinically before lung transplantation. The significance of these and our data is unclear. Is the presence of plexiform arterial lesions in fibrotic ILD an indicator a causal role for the pulmonary arterial vasculopathy in clinical ILD disease progression or if simply a morphological epimarker of the irreversible parenchymal distortion seen in end-stage fibrotic ILD?22 24–26 We speculate about possible explanations:
Geographic and temporal heterogeneity in pulmonary arterial vascular changes throughout the lung. Since the geographic distribution of parenchymal pathology is uneven within the lung, there might be significant heterogeneity in the distribution and severity of the vascular changes. In some patients, we may have found signs of severe local vasculopathy that had yet to cause a significant effect on the PVR as measured in RHC. This idea is supported by previous findings in iPAH histopathology regarding notable heterogeneity in the number and distribution patterns of plexiform lesions within a given PAH lung and between patients with PAH. This occurred without relation to other parameters of vascular remodelling or haemodynamics.11 25 Even if true, this does not account for advanced arterial vasculopathy and of plexiform lesions as a response to chronic hypoxia. Chronic hypoxic changes have been described to cause intimal fibrosis and medial and adventitial thickening, but not higher-grade vascular remodelling including plexiform lesions.27
Shared pathogenic mechanisms between fibrosis and pulmonary arterial vasculopathy. Many mediators including endothelin, VEGF (vascular endothelial growth factor), TGF-β (transfroming growth factor β), NO (nitric oxide), PGE2 (prostaglandin E2), BMPR2 (bone morphogenic protein receptor type 2) and IL-6 (interleukin 6) have been shown to play important roles in both the progression of fibrosis and the development of PH.28 The possibility of shared pathogenic mechanisms between IPF and PH was also demonstrated through microarray gene expression; patients with IPF patients, like patients with PH show an exaggerated expression of mediators of SMC and EC proliferation compared with controls, suggesting the existence of preclinical pulmonary vascular disease.29 The patients end stage fibrotic lung disease theoretically might have caused vascular changes unrelated to clinically measured PH by mPAP in RHC. In other words, the presence of the severe vascular changes might represent end stage lung disease rather than severe PH.
Interestingly, patients who were treated with antifibrotics before lung transplant had significantly lower mPAP and PVR. The role of pirfenidone and nintedanib in patients with IPF and PH is not clear since no data on these agents are currently available. Tyrosine kinase inhibitors such as Imatinib and nintedanib have shown to improve exercise capacity and haemodynamics in patients with advanced WHO group 1 PAH. Pirfenidone which combines antiproliferative and anti-inflammatory activity may have similar effects. However, no data currently support this hypothesis.30
There is no known effective therapy to prevent or treat PH in fibrotic ILD and treatment with pulmonary vasodilators is not recommended based on multiple clinical trials.31–44 Further molecular, biochemical and clinical characterisation subgroup of patients with ILD and high-grade vascular changes might have an important mechanistic, clinical and therapeutic significance. Clinical data including directly measured pulmonary haemodynamics failed to indicate patients at risk for pulmonary arterial vascular pathology. This highlights the challenges in identification of patients with fibrotic ILD who could potentially benefit from future therapeutic interventions.
Our study has several limitations. First, this is a retrospective study that has inherent limitations in patient selection. Second, despite our relatively large sample size as compared with other studies with explanted lungs, the size is still small with inherently reduced statistical power. Third, sampling error is possible and may underestimate the degree of significant vascular changes, especially with disease that is patchy in nature; however, we also view this as strength of our research as this may explain why plexiform pulmonary arterial vasculopathy has yet been better described in end stage ILD.
In summary, we found significant pulmonary arterial vasculopathy in patients with end stage ILD. The vasculopathy did not correlate with the clinical severity of the underlying lung disease and did not correlate with the presence and/or severity of PH as measured by RHC. We found a significant number of patients with end stage lung disease (21%) with plexiform lesions which are traditionally a hallmark of WHO group 1 PAH. The observed vasculopathy might be derived from severe and prolonged hypoxia in the complex biochemical environment of the diseased lung that involve mechanisms similar to those seen with WHO group 1 PAH. These findings indicate that advanced pulmonary arteriopathy is common and may develop in a heterogeneous regional pattern in advanced lung disease prior to the clinical detection of PH. Larger scales studies are needed to confirm our findings and conclusions.
References
Footnotes
Contributors YD collected the data and is the guarantor of the article, taking responsibility for the integrity of the work as a whole from inception to published article. AG collected the data. JS and GD helped write the manuscript. HW, AA and BC reviewed the pathology studies. HZ conducted the statistical analysis. CD reviewed the imaging studies. NM, FCC, GC and AJM were the transplant pulmonologists for the patients included in the study, conducted the pretransplantation evaluation and treated the patients before and after lung transplantation. AJM was a primary coauthor, responsible for the study concept and helped write the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information. All data are deidentified participant data. Data are available on reasonable request from the corresponding author, Dr. Yaniv Dotan, at yaniv.dotan@sluhn.org.