Article Text

Abstract
600 patients aged ≥18 years will be randomised in a 1:1 ratio to nintedanib or placebo. Patients with diagnosis of IPF will be excluded. The study population will be enriched with two-thirds having a usual interstitial pneumonia-like pattern on HRCT. The primary endpoint is the annual rate of decline in forced vital capacity over 52 weeks. The main secondary endpoints are the absolute change from baseline in King’s Brief Interstitial Lung Disease Questionnaire total score, time to first acute interstitial lung disease exacerbation or death and time to all-cause mortality over 52 weeks.
Ethics and dissemination The trial is conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Tripartite Guideline for Good Clinical Practice (GCP) and Japanese GCP regulations.
Trial registration number NCT02999178.
- interstitial fibrosis
- rare lung diseases
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
The term interstitial lung disease (ILD) encompasses a large group of over 200 pulmonary disorders, most of which are classified as rare.1 While idiopathic pulmonary fibrosis (IPF) is the classic fibrosing ILD, clinical data suggest that there is a larger group of patients with differing clinical ILD diagnoses who develop a progressive fibrosing phenotype during the course of their disease. These patients demonstrate a number of similarities to IPF, with their disease being defined by the presence of progressive pulmonary fibrosis, worsening respiratory symptoms, declining lung function, resistance to immunomodulatory therapies and, ultimately, early mortality.2–5 Our terminology to describe this group is ‘patients with progressive fibrosing interstitial lung disease’ (PF-ILD). The working hypothesis is that the response to lung injury in these patients includes the initial development of fibrosis that becomes progressive, self-sustaining and independent of the original clinical association or trigger. At present, there are no therapies with proven efficacy in this population.
Indeed, the American Thoracic Society (ATS) has emphasised that although advances have been made in the treatment of some forms of lung fibrosis, many treatment gaps remain that can only be addressed through large, multicentre, prospective, randomised clinical trials.6 We postulate that targeted antifibrotic therapy may be effective in slowing disease progression in patients with PF-ILD.
Nintedanib is a small molecule tyrosine kinase inhibitor that is approved for the treatment of IPF in the USA, European Union, Japan and a large number of other countries worldwide. Based on clinical and mechanistic parallels between PF-ILD and IPF, it is anticipated that nintedanib will elicit similar effects in this population, that is, slowing the progression of the disease. This assumption is supported by preclinical data indicating that nintedanib impacts fundamental processes of lung fibrosis and that its antifibrotic activity is independent of the cause of the fibrosing lung disease.7–10
This manuscript describes the design of the PF-ILD study, a phase III clinical trial evaluating the efficacy and safety of nintedanib compared with placebo in adult patients with progressive fibrosing lung disease. As efficacy and safety of nintedanib have already been established in IPF, patients with IPF are excluded.
Methods and analysis
Trial design
The PF-ILD trial (1199.247, NCT02999178) is a multicentre, prospective, randomised, double-blind, placebo-controlled study to investigate the efficacy and safety of nintedanib in patients with progressive fibrosing lung disease over 52 weeks. The study is conducted at specialised referral centres experienced in ILD management in approximately 15 countries of the Americas, Europe and Asia.
It is planned to randomise a total of 600 patients in a 1:1 ratio to either oral nintedanib 150 mg (n=300) or matching placebo (n=300) twice daily. The dose of the study drug may be reduced to 100 mg twice daily or interrupted temporarily to manage adverse events (AEs).
For each patient, the study will consist of two parts: part A and part B (figure 1). The duration of part A will be 52 weeks. Part B will be a variable treatment period beyond 52 weeks. In part B, patients will continue on blinded, randomised treatment (nintedanib or placebo) until the end of the trial or until a reason for treatment withdrawal is met. The blinded trial will end once the last randomised patient reaches the week 52 visit and the benefit-risk profile of the nintedanib treatment over 52 weeks has been assessed (figure 1). If the benefit-risk assessment is deemed positive, all patients will have the option of receiving open-label nintedanib.
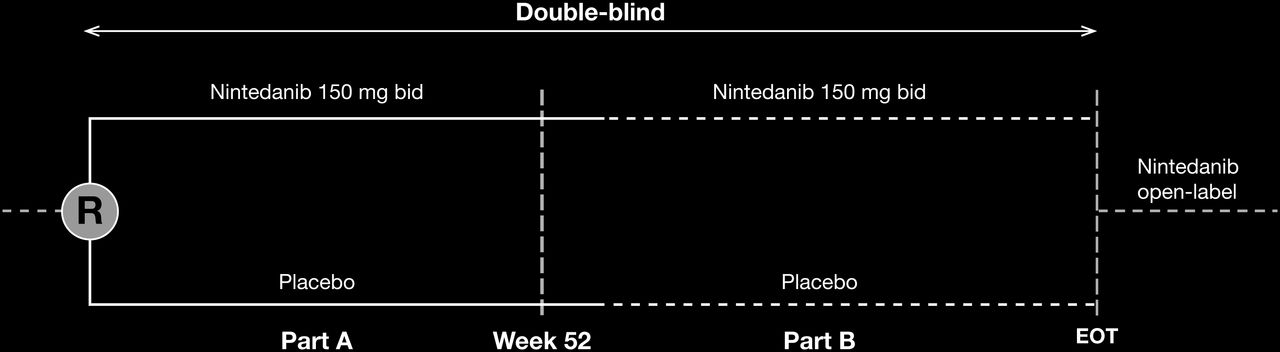
{kind=link}
Trial design. Study design schematic of the PF-ILD trial followed by a separate open-label trial (optional): part A (visits 1 through 9 over 52 weeks) and part B (visit 10 through EOT visit over variable period for each patient). EOT, end of treatment; PF-ILD, progressive fibrosing interstitial lung disease; R, randomisation (1:1 ratio for nintedanib:placebo).
In part A, patients will attend study visits at weeks 2, 4, 6, 12, 18, 24, 30, 36, 44 and 52. Part B visits will take place every 16 weeks. To reduce the amount of missing data, patients who discontinue the trial drug (for any reason) prior to completing the 52-week treatment period will be asked to attend all visits and undergo all examinations as originally planned. In addition, for patients who prematurely discontinue trial medication and are unable to complete the scheduled visits, every attempt will be made to collect information on vital status at week 52 and at the time of data cut-off for the primary data analysis and at the end of the trial.
The study population will be enriched for patients with PF-ILD with usual interstitial pneumonia (UIP)-like pattern on high-resolution CT (HRCT), and they will make up two-thirds of the study population (ie, 400 of the 600 patients enrolled will have a UIP-like pattern). The protocol-defined HRCT criteria (table 1) will be based on those used in the INPULSIS studies, the phase III nintedanib trials in IPF.11 12 Patients meeting these protocol-defined criteria will be referred to as patients with UIP-like fibrotic patterns only; those not meeting the criteria will be referred to as patients with other HRCT fibrotic patterns.
HRCT criteria for UIP-like fibrotic patterns in patients who meet the protocol criteria for PF-ILD*
There will be two coprimary analysis populations: the first will comprise all patients (the overall population); the second will comprise all patients with HRCT with UIP-like fibrotic pattern only. The sample size of 600 patients (300 per treatment group) will provide adequate power to detect a clinically meaningful treatment difference in either of the coprimary populations. Online supplementary table S2 contains the power properties of several scenarios for the treatment effect in the two coprimary populations.
Supplementary file 1
Patient population
Patients aged ≥18 years will be eligible for the trial if they have a physician-diagnosed fibrosing ILD, such as connective tissue disease (CTD)-associated ILD, chronic fibrosing hypersensitivity pneumonitis (HP), idiopathic non-specific interstitial pneumonia (iNSIP), unclassifiable idiopathic interstitial pneumonia (IIP), environmental/occupational lung disease or sarcoidosis, present with features of diffuse fibrosing lung disease of >10% extent on HRCT and meet the protocol criteria for progression within 24 months of screening, as assessed by the investigator. The criteria for evidence of progression are worsening lung function (clinically significant, ≥10% relative decline in forced vital capacity (FVC)) or worsening lung function (≥5–<10% relative decline in FVC) together with worsening respiratory symptoms and/or evidence of increasing fibrosis on chest imaging, despite treatment with unapproved medications used in clinical practice to treat ILD. A full list of the criteria for progression is provided in online supplementary table S1.
At screening, the most recent HRCT image (taken within the previous 12 months) will be evaluated by central review according to the protocol to ensure that relevant lung fibrosis is present. In addition, the HRCT pattern will be determined for randomisation stratification. This review will be performed by two independent specialists in thoracic radiology with extensive expertise in the interpretation of HRCT of the chest. The reviewers will be blinded to patients’ demographic and clinical data.
In addition, eligible patients will have a FVC>45% of predicted value and a diffusing capacity of the lungs for carbon monoxide (DLCO) of >30% and <80% of predicted at randomisation.
As nintedanib is already approved for patients with IPF, patients will be excluded if they meet diagnostic criteria for IPF according to the ATS/European Respiratory Society (ERS)/Japanese Respiratory Society/Latin American Thoracic Association 2011 guidelines.13 Patients with contraindications to nintedanib, as labelled, will also be excluded. Other exclusion criteria include treatment at baseline with any of the following drugs: azathioprine, cyclosporine, tacrolimus, rituximab, cyclophosphamide, mycophenolate mofetil or oral corticosteroids (>20 mg/day). Patients who require use of these medications to control an underlying disease such as CTD will not be considered for participation in the study. In case of worsening of PF-ILD and/or worsening of the underlying CTD during the treatment period, the use of any of these drugs will be allowed after 6 months of study treatment, if judged necessary by the investigator.
A full list of inclusion and exclusion criteria is given in online supplementary table S1.
Study endpoints
As in the INPULSIS trial, the primary endpoint is the annual rate of decline in FVC (mL/year), calculated from measurements obtained over 52 weeks of treatment.11 Spirometry testing will be performed according to ATS/ERS criteria,14 on machines provided by the sponsor. Main secondary endpoints are the absolute change from baseline in King’s Brief Interstitial Lung Disease Questionnaire (K-BILD) total score at week 52, time to first acute ILD exacerbation or death over 52 weeks and time to death over 52 weeks. Other endpoints are shown in table 2.
Study endpoints
For safety analyses, laboratory tests (full blood count, pregnancy test and metabolic panel, including liver function tests) will be performed at each visit, including the end of treatment and follow-up visits. In addition, the protocol provides provision for intermediate laboratory test visits as needed, for additional safety monitoring, at the discretion of the investigator.
Statistical analysis
Efficacy and safety analyses will be conducted using data from patients who are randomised to treatment (nintedanib or placebo) and receive at least one dose of study medication. The primary assessment of benefit risk will be based on efficacy and safety data over 52 weeks. Data collected beyond 52 weeks (ie, during part B) will provide supportive longer term information on the effect of nintedanib on patients with PF-ILD in a controlled manner, especially for early enrollers. Similar to the INPULSIS trials,11 the following models will be used to analyse the data: random coefficient regression (random slopes and intercepts) model for the primary endpoint; mixed effects models for repeated measures for all other continuous secondary endpoints; Cox proportional hazards models and Kaplan-Meier plots for time-to-event secondary endpoints; and logistic regressions for binary secondary endpoints.
Formal statistical testing will be performed using data from the two coprimary populations, that is, the overall population and all patients with HRCT with UIP-like fibrotic pattern only. To maintain an overall type 1 error rate of 5%, a Hochberg procedure15 will be used for multiplicity adjustment.16 Since establishing an effect in either of these two populations is clinically relevant, the trial will be considered positive if it demonstrates efficacy in either or both of the two coprimary populations. For the primary endpoint, statistical significance will be declared if the analyses in both coprimary populations are significant at the two-sided 5% level or if the analyses in either coprimary population are statistically significant at the two-sided 2.5% level. For the subgroup of patients with other HRCT fibrotic patterns, it is not expected that there will be sufficient power to achieve statistical significance. However, results in this subgroup will also be assessed and presented descriptively using the treatment-effect estimate and its CIs. All available data, including data collected after premature discontinuation of trial medication but excluding data after lung transplant, will be used in the analyses of all efficacy endpoints.
Trial organisation and oversight
This trial will be guided by a steering committee consisting of clinical experts in ILD and representatives of the sponsor, Boehringer Ingelheim (see online supplementary table S3). An independent data monitoring committee will perform regular review of the data, particularly those relating to serious AEs, or AEs leading to discontinuation of study drug and laboratory parameters. An independent adjudication committee will review medical documentation for all fatal cases and adjudicate all deaths to either cardiac, respiratory or other causes and will assess all AEs categorised as major adverse cardiovascular events.
The trial will be carried out in compliance with the ethical principles of the Declaration of Helsinki and in accordance with the International Conference on Harmonisation Tripartite Guideline for Good Clinical Practice (GCP) and Japanese GCP regulations (Ministry of Health and Welfare Ordinance No. 28; 27 March 1997).
All relevant documentations were approved by the respective institutional review board or independent ethics committee and competent authority before the start of the study. Written, informed consent will be obtained from all patients before they are enrolled in the study.
Discussion
Rationale for conducting the trial
The PF-ILD study is an innovative phase III trial evaluating the antifibrotic agent nintedanib in patients with fibrosing ILD who, independent of their original clinical classification, exhibit a progressive fibrosing phenotype. In this group of patients with PF-ILD, the natural history appears to follow a course similar to IPF, with worsening of respiratory symptoms, lung function, health-related quality of life (HRQoL) and functional status and with early mortality, despite treatment with currently available non-approved immunomodulatory therapies. With the exception of nintedanib and pirfenidone, which are available only for patients with IPF, there is no approved therapy for patients with PF-ILD. Separate from IPF and systemic sclerosis-associated ILD, no prospective, controlled clinical trials have been performed in other forms of fibrosing ILDs, probably due to their relative rarity.
From the authors’ experience and based on limited literature available,17 18 within each fibrosing ILD subgroup (eg, iNSIP or chronic HP), the number of patients is similar to, or lower than, the number with IPF; the number of patients with a progressive phenotype within each group, while still significant, is even lower. Therefore, we suggest that grouping patients with PF-ILD together is justified based on their clinical and biological similarities and is also the only feasible way to conduct an appropriately powered prospective clinical study to potentially provide efficacious therapy for these patients.3
Rationale for patient selection
According to the original clinical ILD diagnosis, the study population is expected to include mainly patients with physician diagnosis of the following: iNSIP; unclassifiable IIP; CTD-ILD (primarily rheumatoid arthritis-ILD); chronic HP; or environmental/occupational fibrosing lung diseases.
The protocol criteria for patient selection mirror the physician’s assessment in clinical practice, as they are based on a composite of the three clinically recognised dimensions of disease progression: worsening of symptoms, worsening of lung function and increasing fibrosis on chest images. To date, there is no universal consensus on how worsening of symptoms should be measured, what represents a clinically meaningful threshold decline in FVC and how the extent of fibrosis should be quantified using serial chest images in the real-life setting. Accordingly, the decision whether the ILD of an individual patient with pulmonary fibrosis is progressing and requires a change in treatment is made by the treating physician without the support of applicable treatment guidelines. It is anticipated that the majority of patients will be included in the study based on a relative decline in FVC (per cent predicted) ≥5 to <10% within the last 24 months, combined with worsening of symptoms and/or qualitative worsening of disease extent on chest imaging despite treatment with unapproved medication used in clinical practice to treat ILD. This requirement is aimed at supporting the inclusion of patients with ILDs at the stage when the underlying pathology is predominantly fibrotic and clinically progressive. It is understood that for a number of ILDs (eg, asbestosis, some cases of fibrotic HP), resistance to anti-inflammatory/immunomodulatory therapy is expected and therefore only non-pharmacological treatment is used. These cases are also allowed.
Additional inclusion criteria such as >10% extent of fibrosing lung disease on HRCT and DLCO <80% predicted will be used to aid in the selection of patients with a progressive phenotype.
Rationale for study endpoints
The study endpoints are consistent with previous IPF trials and reflect clinically meaningful outcomes. FVC is an established efficacy parameter and mean changes over time are considered relevant to assess the effect of a pharmacological intervention at population level.19–22 The use of FVC as a primary endpoint served as the basis for worldwide regulatory approvals for nintedanib and pirfenidone in IPF. Similar to IPF, the accelerated decline in lung function in other ILDs over time is considered consistent with disease progression and is thought to be associated with mortality.19 23–30 Based on these, the annual rate of decline in FVC was selected as primary endpoint of the study.
PF-ILD places a significant burden on daily life and has a severe impact on how patients feel and function. Selection of patient-reported outcome measures was based on two documents: the US Food and Drug Administration (FDA) reports ‘The Voice of the Patient,’31 and a manuscript from the Outcome Measures in Rheumatology working group.32 Both documents conclude that the most relevant aspects of the disease for patients with IPF, and with ILDs in general, are HRQoL, dyspnoea and cough. Accordingly, the absolute change from baseline to week 52 in the K-BILD total score has been included as one of the main secondary endpoints. This health status questionnaire was developed in multiple languages specifically for patients with ILD.33–36
To further support the potential effect of nintedanib on slowing disease progression, time to death over 52 weeks will be evaluated as another main secondary endpoint. In clinical trials in IPF with treatment durations between 12 and 18 months, the rate of all-cause mortality has been shown to be relatively low37; the mortality rate in patients with PF-ILD is anticipated to be similar. The PF-ILD study is powered for the primary endpoint, the annual rate of decline in FVC, with a total sample size of 600 patients, and hence, it is not powered to demonstrate a statistically significant reduction in mortality over 52 weeks. However, inclusion of all-cause mortality as a main secondary endpoint will provide further clinically meaningful information on the effect of nintedanib on patients with progressive fibrosing ILD. These data will be complemented by the additional main secondary endpoint, time to first acute ILD exacerbation or death over 52 weeks.
Rationale for comedication restrictions
Immunomodulatory medications, that is, azathioprine, cyclosporine, tacrolimus, rituximab, cyclophosphamide, mycophenolate mofetil and oral corticosteroids, are often used in clinical practice to manage ILD, although their benefit-risk profiles in ILD have not been established and they are not approved for the treatment of ILD in most countries. In addition, these drugs are less frequently used and/or are used off-label in the treatment of CTDs. Lastly, there are no data available on the concomitant use of these medications with nintedanib. Therefore, to avoid the potential confounding impact of these drugs on the assessment of the safety and efficacy of nintedanib in patients with PF-ILD, their use will not be allowed at randomisation and during the first 6 months of the treatment period. As the protocol requires that eligible patients progress despite treatment with these medications (ie, do not, or no longer, benefit from these drugs), prohibition of these medications is considered justified for the purposes of this study. Initiation of concomitant immunomodulatory treatment will be allowed after 6 months of study treatment to manage worsening of either the ILD or underlying CTD, as judged by the investigator.
Rationale for enrichment design
Currently available data suggest that the ILD injury pattern seen on chest imaging or by histopathology has a major impact on prognosis. The prognosis and clinical behaviour of fibrosing ILDs, particularly those with a UIP pattern, appear to be similar to those of IPF. We hypothesise that nintedanib will have a similar relative effect on lung function decline in patients with PF-ILD as in patients with IPF, that is, a reduction of approximately 50% in the annual rate of decline in FVC. However, it is expected that the absolute difference between the treatment groups will be larger in patients with PF-ILD with UIP-like fibrotic pattern since those patients seem to have a larger annual rate of decline than patients with other HRCT fibrotic patterns. Based on these assumptions, an enrichment design will be used to increase the likelihood of detecting a clinically meaningful effect of nintedanib with the greatest statistical efficiency, therefore ensuring feasibility of the enrolment of patients with this rare disease. This approach is consistent with the prognostic enrichment strategies in the draft FDA Guidance for Industry: Enrichment Strategies for Clinical Trials to Support Approval of Human Drug and Biological Products.38 This guidance document specifies that the use of prognostic enrichment strategies in the investigation of drugs that are intended to delay progression can be considered in order to increase the absolute effect size, generally allowing for a smaller sample size, as long as the relative effect size is considered to be similar between subpopulations.
Conclusions
This is the first innovative study in which patients with a variety of clinical diagnoses of fibrosing ILD are grouped together based on the similarity of their biological and clinical behaviour. It will explore the efficacy and safety of nintedanib in a group of patients with PF-ILD and will further our understanding of the natural history of fibrosing ILDs.
Acknowledgments
The authors wish to acknowledge the contribution of David M Hansell to the design of this trial and the development of this manuscript. Medical writing support was provided by Lauren Liss, PhD (Boehringer Ingelheim Pharma GmbH & Co. KG) and Nicky French, PhD (on behalf of Complete HealthVizion, which was contracted and compensated by Boehringer Ingelheim Pharma GmbH & Co. KG).
References
Footnotes
Contributors KRF, KKB, AUW, ECB, HRC, VC, AD, YI, LR, SW and RSH are members of the Trial Steering Committee, responsible for the design of the PF-ILD trial. WM also provided input into the trial design and the content of this manuscript. FLM and HS are the trial statisticians, responsible for the statistical elements of the trial design. All authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors. They take full responsibility for the scope, direction, content and editorial decisions relating to the manuscript, were involved in all stages of development and have approved the submitted manuscript.
Funding The PF-ILD trial is funded by Boehringer Ingelheim Pharma GmbH & Co. KG. The authors received no compensation related to the development of the manuscript.
Competing interests KRF reports grants and personal fees from Genentech, grants from Afferent, and personal fees from Boehringer Ingelheim, Veracyte, Roche, Biogen, Aeolus and Pharmakea. KKB reports grants from NHLBI, grants and personal fees from Actelion, Amgen, Gilead, and personal fees from Almiral, Altitude Pharma, Astra Zeneca, Byer, Biogen/Stromedix, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Centocor, Fibrogen, Galecto, GlaxoSmithKline, MedImmune, Novartis, Pfizer, Promedior, Roche/Genentech, Sanofi/Genzyme, Veracyte, Aeolus and ProMetic. AUW reports lecturing and consultancy fees from Boehringer Ingelheim, Roche, InterMune, Actelion and Bayer. HRC reports personal fees from MedImmune, Bayer, Biogen, Boehringer Ingelheim, Xfibra, Genoa, Gilead, GlaxoSmithKline, Mesoblast, Moerae Matrix, PharmAkea, Promedior, Prometic, Pulmatrix, Pulmonary Fibrosis Foundation (senior medical advisor), Unity, Aeolus, aTyr Pharmaceuticals, Pfizer, UCB Celltech, GBT, Veracyte, Patara, Samumed, Alkermes, Five Prime and Takeda. VC reports personal fees from Actelion, Bayer, Biogen Idec, Boehringer Ingelheim, Gilead, GSK, Intermune, MSD, Novartis, Pfizer, Roche and Sanofi, grants from Actelion, Boehringer Ingelheim, GSK, Pfizer and Roche, and personal fees from Boehringer Ingelheim. YI reports lecture and advisory fees from Boehringer Ingelheim, Shionogi & Co., Ltd., Takeda, Nobel Pharma, Novartis, Serendex, Bayer, Chugai and Daiichi Sankyo. LR reports grants and personal fees from InterMune, and personal fees from MedImmune, Biogen, SanofiAventis, Roche, Takeda, ImmuneWorks, Shionogi, Boehringer Ingelheim and Pliant Therapeutics. ECB, HS, WM and RSH are full-time employees of Boehringer Ingelheim. FLM was a full-time employee of Boehringer Ingelheim at the time of manuscript development. SW and AD report no competing interests.
Ethics approval The trial is conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Tripartite Guideline for Good Clinical Practice (GCP) and Japanese GCP regulations.
Provenance and peer review Not commissioned; externally peer reviewed.