Article Text

Abstract
Introduction In adult asthma, combination inhaled corticosteroid (ICS)/fast-onset long-acting beta agonist (LABA) used solely as reliever therapy may represent an effective and safe alternative to ICS maintenance and short-acting beta agonist (SABA) reliever therapy.
Objective To compare the efficacy and safety of ICS/fast-onset LABA reliever therapy with ICS maintenance and SABA reliever therapy in adults with asthma.
Methods and analysis A 52-week, open-label, parallel group, multicentre, phase III randomised controlled trial with 1:1 randomisation to either budesonide/formoterol Turbuhaler 200/6 µg, one actuation as required for symptom relief, or budesonide Turbuhaler 200 µg, one actuation twice daily and terbutaline Turbuhaler 250 µg, two actuations as required for symptom relief. 890 adults aged 18–75 years with asthma for whom maintenance ICS and SABA reliever therapy is indicated by current guidelines will be recruited in New Zealand. The primary outcome variable is the rate of severe exacerbations per patient per year. This study will investigate a novel treatment regimen that might lead to a paradigm shift in asthma management for adults for whom guidelines currently recommend maintenance ICS and SABA reliever therapy.
Ethics and dissemination Ethical approval has been granted (15/NTB/178). Study findings will be published according to Iinternational Committee of Medical Journal Editors' recommendations.
Trial registration number ACTRN12616000377437; Pre-results.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Asthma is a major health problem globally.1 Most adults with asthma have so-called ‘intermittent’ or mild disease2; however, the evidence base for their management is less well established than for moderate to severe asthma. Treatment is usually initiated with a short-acting beta agonist (SABA) for symptom relief; however, there are concerns regarding the potential risks associated with such use long term, and in the setting of a severe exacerbation.3 4
Inhaled corticosteroid (ICS) therapy reduces morbidity and mortality in asthma,5 and this is the basis of past guideline recommendations to use regular ICS for patients using their SABA on more than two occasions in a week.6 However, the 2014 Global INitiative for Asthma (GINA) report7 expanded the indication for regular low-dose ICS to include patients with symptoms on two occasions or more a month, based on the lack of evidence for safety of treatment with SABA alone and the findings of the START study.8 9 Nevertheless, surveys of patients with asthma show many are reluctant to take ICS every day,2 10 11 which is not surprising as patients are required to take regular twice daily treatment regardless of whether they have symptoms. Recognition by primary care practitioners that such patients are unlikely to be adherent with regular ICS treatment is likely to contribute to their reluctance to prescribe ICS. These limitations have led to the recognition that alternative regimens to maintenance ICS and SABA reliever therapy are required.
One alternative regimen is combination ICS/fast-onset long-acting beta agonist (LABA) reliever therapy. This has major potential advantages of allowing titration of ICS therapy according to need, and of increasing ICS usage by patients who would otherwise be poorly adherent and over-rely on their SABA.12–14 It also enables ICS to be delivered early in the course of worsening asthma, which is more effective in preventing severe exacerbations than increasing SABA alone,15 and it enables repeated doses of ICS to be delivered by patients with severe exacerbations of asthma. This has been reported in at least one large trial to lead to at least as much clinical benefit as systemic corticosteroids in this clinical situation.16
Clinical evidence in support of the combination ICS/fast-onset LABA reliever therapy regimen comes from randomised controlled trials (RCT) such as the Beclomethasone plus Salbutamol Treatment study, in which a symptom-based ICS/SABA combination inhaler was shown to be more effective in reducing exacerbations than SABA reliever therapy in patients with mild asthma.17 The symptom-driven as-required use of combination ICS/SABA in a single inhaler had equivalent efficacy to regular ICS treatment in this population. In a study of different ICS regimens, symptom-based use of both ICS and SABA (in separate inhalers) had similar efficacy to a physician-based strategy of six weekly adjustment of maintenance ICS dose in addition to SABA reliever use, and greater efficacy during high-risk periods of increased viral infections and allergen exposures.18 The greater efficacy of ICS/fast-onset LABA reliever therapy than either a SABA or fast-onset LABA as reliever therapy has been demonstrated from studies of the Maintenance and Reliever Therapy regimen, in which patients take the same combination ICS/fast-onset LABA inhaler for both maintenance and reliever therapy.15 19 In patients with infrequent symptoms but elevated fraction of exhaled nitric oxide (FeNO) at baseline, a symptom-based ICS/fast-onset LABA combination inhaler led to reduced FeNO, an important biomarker of airway inflammation, compared with fast-onset LABA reliever therapy alone, indicating better control of airway inflammation.20
Based on this evidence, two double-blind regulatory RCTs of as-needed budesonide/formoterol (SYmbicort Given as needed in Mild Asthma, SYGMA) are currently underway,21 together with an open-label RCT of as-needed budesonide/formoterol in patients currently taking only SABA reliever therapy (Novel Symbicort Therapy As Reliever Therapy, Novel START).22 To investigate the efficacy and safety of budesonide/formoterol ICS/fast-onset LABA as sole reliever therapy in a broader population of patients for whom ICS is currently indicated, the PRACTICAL (PeRsonalised Asthma Combination Therapy with an Inhaled Corticosteroid And fast-onset Long acting beta agonist) trial will therefore be undertaken (ACTRN12616000377437). The PRACTICAL study is an open-label study funded by the government-funded Health Research Council of New Zealand and has undergone full external peer review as part of the funding process.
Methods
Objectives
The primary objective is to compare the efficacy of the ICS/fast-onset LABA reliever therapy regimen with the maintenance ICS and SABA reliever therapy regimen in patients in whom maintenance ICS and SABA reliever therapy is recommended.
The secondary objectives include the following:
to compare the safety of the ICS/fast-onset LABA reliever therapy regimen with the ICS maintenance and SABA reliever therapy regimen
to determine whether baseline clinical and socioeconomic characteristics such as reported beta agonist use, type 2 immune response profile, smoking status, history of severe exacerbations, deprivation index or housing condition predict preferential response to randomised treatments
to examine patterns of inhaler use with the randomised treatments
to examine the cost-effectiveness of the randomised treatments
to examine patient attitudes to the treatment regimens.
Design
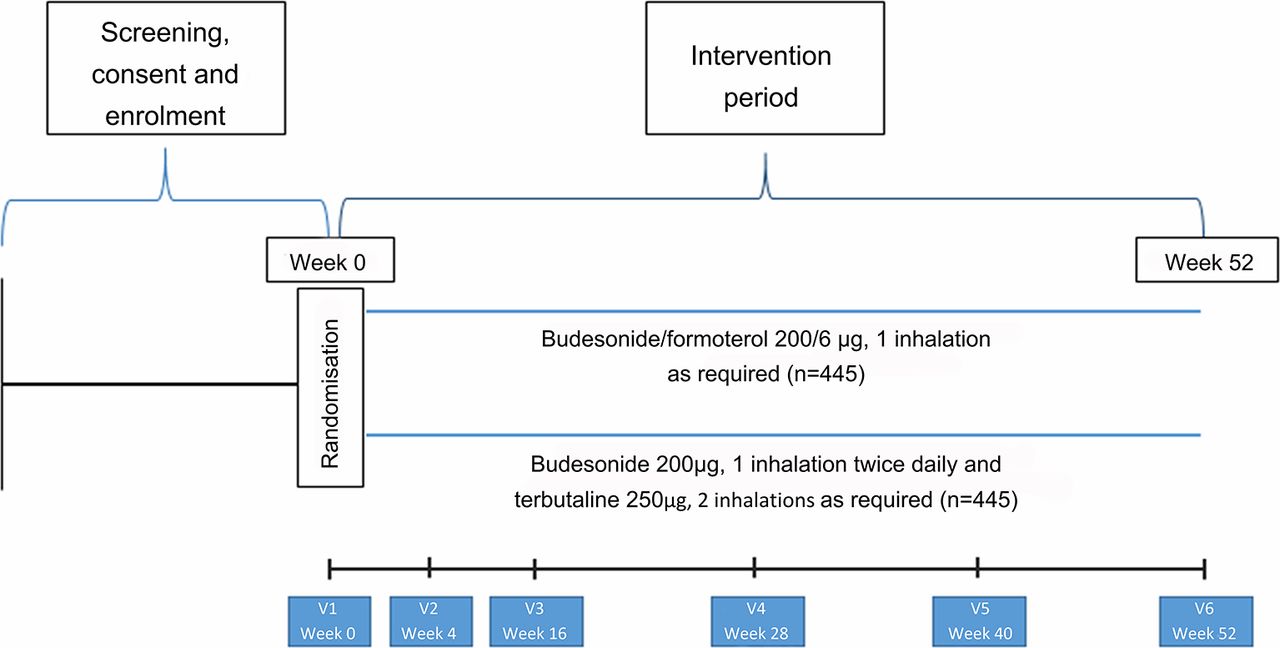
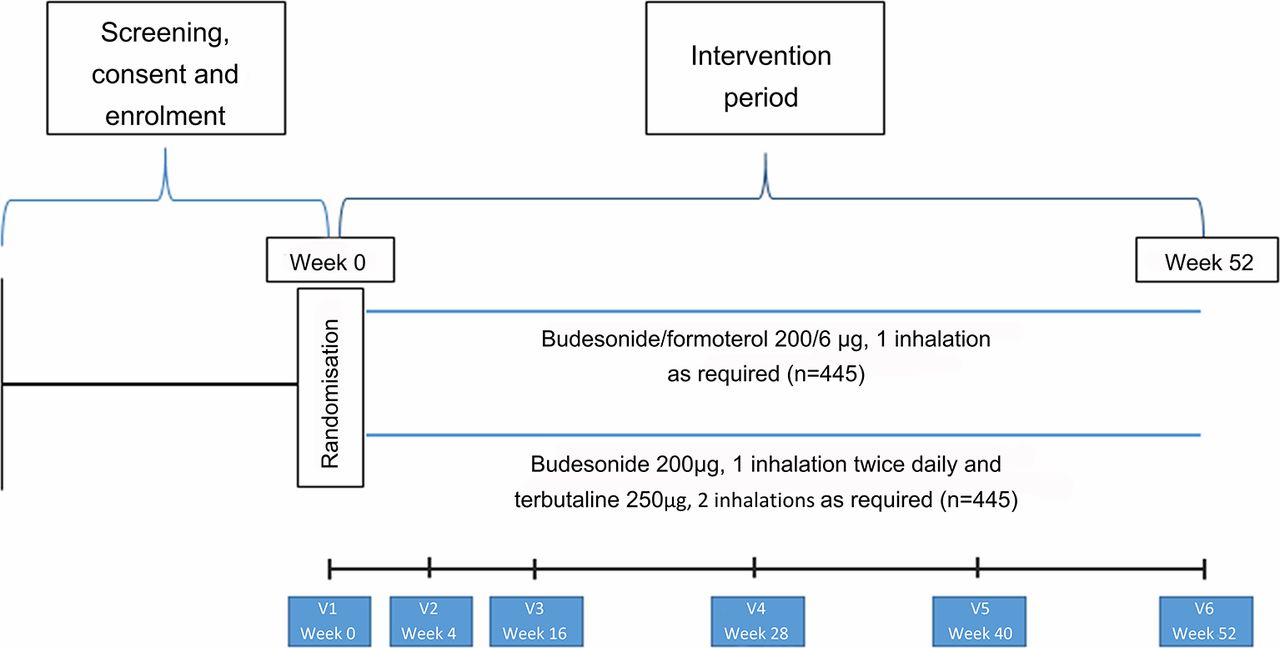
The study is a 52-week, open-label, parallel group, multicentre, RCT that will be performed in New Zealand. The clinical trial will compare the efficacy and safety of two asthma treatment regimens: budesonide/formoterol Turbuhaler taken as required for relief of symptoms (ICS/LABA reliever therapy), and regular twice-daily budesonide Turbuhaler together with terbutaline Turbuhaler taken as required for relief of symptoms (maintenance ICS and SABA reliever therapy).
Participants
A total of 890 adult patients with doctor-diagnosed asthma, using as-needed SABA with or without low-dose or moderate-dose ICS, will be recruited in accordance with the inclusion and exclusion criteria shown in table 1. All participants will sign written informed consent. The study has been approved by the Northern B Health Disability and Ethics Committee, Ethics number 15/NTB/178.
Inclusion and exclusion criteria
Recruitment sites
Participants will be recruited from the Medical Research Institute of New Zealand and primary care-based research centres in New Zealand.
Randomised treatments
Participants will be randomised in equal proportions to one of two treatments:
ICS/LABA reliever therapy: budesonide/formoterol Turbuhaler 200/6 µg, one inhalation for relief of symptoms as required
maintenance ICS and SABA reliever therapy: budesonide Turbuhaler 200 µg, one inhalation twice daily, and terbutaline Turbuhaler 250 µg two inhalations for relief of symptoms as required.
All participants will be given education on medication use and inhaler technique, and a written asthma self-management (‘action’) plan relating to their randomised group. It is recognised that issuing each participant with an asthma management plan may improve asthma control and reduce the number of exacerbations. Given that issuing all asthmatics with a personalised asthma management plan is a tenet of all asthma guidelines, it was felt that this was a mandatory part of standard care within the study protocol. All participants will receive an action plan so this should affect both groups equally.
If the participant has an exacerbation during the study, they will be asked to contact their general practitioner (GP) for assessment and management or to visit an emergency department (ED) or after-hours clinic as outlined in their self-management plan.
Randomisation
Participants will be block-randomised. Randomisation will be stratified by site and by whether participants used ICS therapy prior to enrolment. A computer-generated randomisation sequence will be generated by the study statistician, independent of the investigators undertaking recruitment and subsequent visits.
Allocation concealment and blinding
Allocation concealment will be by a secure database that contains the randomisation sequence. A participant’s treatment allocation will only be revealed to the researchers when that participant is randomised via the electronic clinical record form (eCRF). There is no blinding to allocated intervention in this study. Study investigators, study staff and participants will be aware of the treatment allocation. However, the study statistician will be blinded while performing the analysis of the primary outcome variable.
Clinic visits
Participant flow and interventions are shown in figure 1 and table 2. Participants will be seen for the initial visit and 4, 16, 28, 40 and 52 weeks after randomisation. Participants will be issued with up to five new Turbuhalers at each visit (depending on the randomised treatment, time to the next visit and medication use during the previous treatment period). At each visit participants will complete the Five-Item Asthma Control Questionnaire (ACQ-5).23 Measurements of forced expiratory volume in 1 s and forced vital capacity and of FeNO will be made at weeks 0, 16 and 52, and peripheral blood eosinophil count and serum periostin (at selected sites) at week 0. At each visit participants will be instructed on their randomised medication use, and the accompanying self-management plan, as outlined above. Full details of study procedures are given in the formal full protocol document provided as an online supplementary file.
{kind=link}
Participant flow throughout the study.
Visit overview
Electronic monitoring substudy
A total of 110 participants will have electronic monitors attached to each Turbuhaler device to record the date and time of every actuation, to allow a detailed assessment of the patterns of use of the randomised treatments.
Electronic monitoring devices will have individual identification numbers and will remain patient-specific during the course of the study. A comprehensive trial quality control programme will be implemented in which all monitors are tested prior to dispensing and during the full study period, as previously.24 25 The accuracy of the electronic monitors in detecting Turbuhaler medication use has been confirmed in a bench study.26 Electronic data on days of study visits will be removed prior to analysis, because dose-dumping may occur at this time. The electronic database will be programmed not to count repeat actuations within 3 s.
Outcome measures
Outcome measures for the main study are shown in box 1 and for the electronic monitoring substudy in box 2. The primary outcome measure is the severe asthma exacerbation rate expressed as number of severe exacerbations per patient per year. A severe asthma exacerbation is defined according to the American Thoracic Society/European Respiratory Society (ATS/ERS) criteria27:
the use of systemic corticosteroids for at least 3 days, or
hospitalisation or ED visit because of asthma, requiring systemic corticosteroids.
Study outcome measures
Primary outcome
Severe asthma exacerbation rate expressed as number of severe exacerbations per patient per year. A severe asthma exacerbation is defined according to the ATS/ERS criteria:
The use of systemic corticosteroids for at least 3 days because of asthma, or
Hospitalisation or ED visit because of asthma, requiring systemic corticosteroids
Secondary outcomes
Clinical outcomes:
Time to first severe exacerbation of asthma
The proportion of severe asthma exacerbations defined by each of (1) and (2) above
The proportion of participants with at least one severe exacerbation
Five-Item Asthma Control Questionnaire score
On-treatment FEV1 (ie, participants will not be required to withhold study medications prior to visits)27
On-treatment FEV1 percentage predicted
FeNO (a measure of type 2 immune response airways inflammation)
Proportion of participants withdrawn and reason
Adverse events:
Adverse events
Serious adverse events
Cost-effectiveness:
The medical costs (medications, emergency medical and ED visits, hospital admissions) and non-medical costs (days off work) will be calculated for each treatment regimen. The cost-effectiveness data collected will allow extrapolation to future pricing models with lower cost generic medications.
Work Productivity and Activity Impairment questionnaire: asthma
Patient attitudes:
Beliefs about medicines questionnaire28
ATS/ERS, American Thoracic Society/European Respiratory Society; ED, emergency department; FeNO, nitric oxide; FEV1, forced expiratory volume in 1 s.
Electronic monitoring substudy outcome measures
Primary outcome
Mean inhaled corticosteroid (ICS) dose per day (budesonide µg/day)
Secondary outcomes
Inhaled corticosteroid (ICS) use:
Proportion of participants with at least 1 day of no ICS use
Number of days of no ICS use
Number of ≥7 consecutive day periods of no ICS use
Number of ≥14 consecutive day periods of no ICS use
Longest duration of no ICS use (days)
Corticosteroid use:
Total oral corticosteroid dose
Number of courses of oral corticosteroid per year
Composite systemic corticosteroid exposure/year in which the total ICS dose/year, converted to oral prednisone-equivalent dose for systemic effects on adrenal function, is added to the oral prednisone dose per year, as previously defined (budesonide 5000 µg inhaled equivalent to prednisone 10 mg oral). For other systemic corticosteroids, conversion to prednisone-equivalent doses will be undertaken by reference to the British National Formulary.29
High beta agonist use, defined as >16 actuations of terbutaline or >8 actuations of budesonide/formoterol per 24-hour period
Proportion of participants with at least one episode of high use
Number of days of high use
Number of days of high use in participants with at least 1 day of high use
Number of high beta agonist use episodes without medical review in the following 48-hour period, 7-day period and 14-day period in participants who had at least one high beta agonist use episode, as previously defined
Proportion of high use episodes without medical review within 48 hours, 7 days and 14 days
Marked beta agonist overuse, defined as >24 actuations of terbutaline or >12 actuations of budesonide/formoterol per 24-hour period, as previously defined19
Proportion of participants with at least one episode of marked overuse
Number of days of marked overuse
Number of days of marked use in participants with at least 1 day of marked overuse
Number of marked beta agonist use episodes without medical review in the following 48-hour period, 7-day period and 14-day period in participants who had at least one marked beta agonist use episode
Proportion of marked use episodes without medical review within 48 hours, 7 days and 14 days
Maximum number of beta agonist actuations in a 24-hour period
Use of study medications in the 14 days prior to severe exacerbations, as previously defined,30 with graphical presentation of the median (IQR) daily medication use for the randomised groups, and the medication use for the individual participants within each randomised group
Hospital admissions will be verified with the Ministry of Health for the year prior to and the year of enrolment in the study. Participants are specifically questioned regarding unscheduled contact with health services due to a deterioration in asthma control (hospital, ED, GP, after hours) in detail at each visit. Investigators will obtain documentation confirming any severe exacerbations. Each participant confirms at the point of randomisation that they do not have home supply of steroids for use in worsening asthma so self-medication is unlikely to be a significant issue.
Secondary outcome measures have been chosen to provide information on the safety of the randomised treatments, as well as the effect on asthma control, level of airway obstruction and type 2 immune response-type airway inflammation. In addition health economic modelling will be performed to compare the costs of the two approaches, both in terms of direct medical costs and indirect non-medical costs. The full analysis, including the health economic analysis, will be prespecified prior to the completion of data collection.
The New Zealand Deprivation Index is a national small-area index of socioeconomic deprivation derived from census data. A New Zealand Deprivation Index score will be assigned to each participant and combined with housing, quality of life, employment status and occupation data and measures of presenteeism and absenteeism, allowing a more granular investigation of the influence of socioeconomic status.
Sample size calculation
The primary outcome variable is the rate of severe exacerbations per patient per year. Assuming a dropout rate of 10%, 890 patients will be recruited to enable a sample size of 400 completed patients in each treatment arm, resulting in 90% power, alpha of 5%, to detect a 38% reduction in the rate of severe exacerbations from 0.30 to 0.185 per year.
The primary outcome variable for the substudy is the mean ICS dose per day. Assuming a dropout rate of 10%, 110 patients will be recruited into the substudy to ensure a sample size of 50 completed patients in each treatment arm, resulting in 90% power, alpha of 5%, to detect a 18% decrease in ICS use (μg/day) with ICS/LABA reliever therapy, compared with 264 µg/day in the standard ICS and SABA regimen. This calculation is based on data from our previous study of ICS adherence in stable at-risk patients prescribed regular budesonide/formoterol in which participants took a mean (SD) 66% (27) of their prescribed ICS dose.31
Statistical analysis
The statistical analysis will be by ‘intention to treat’, testing the hypothesis that as-needed budesonide/formoterol is superior to low-dose ICS with as-needed SABA for reducing severe exacerbations. The primary analysis is comparison of the rate of severe exacerbations per patient per year by Poisson regression with an offset for the days of observation and a fixed effect for baseline frequency of SABA use and self-reported number of prior severe exacerbations in the year before recruitment. Overdispersion will be evaluated prior to analysis and a corrected analysis applied if necessary. A sensitivity analysis will include the following potentially important predictors of response: age, sex, ethnicity, smoking status, baseline ACQ-5 score, severe exacerbation in the previous year, baseline ICS use, baseline FeNO, site and baseline blood eosinophil count. This will account for different distributions of these variables in the treatment groups and to increase precision of the estimates of differences. Survival analysis with Kaplan-Meier plots and Cox’s proportional hazards will be used to calculate the HR for the time to first exacerbation. Details of planned secondary and subgroup analyses can be found in the full protocol, provided as an online supplementary file. SAS V.9.4 will be used.
Data Safety Monitoring Committee
A Data Safety Monitoring Committee (DSMC) will be established, which is independent from the study team. The DSMC will review all serious adverse events and the results of the interim safety statistical analysis undertaken when 500 patients have been randomised. The interim safety statistical analysis will be conducted by the study statistician, Professor Mark Weatherall, for all asthma-related hospitalisations because of asthma and requiring systemic corticosteroids. This analysis will be performed masked to treatment allocation (the trends for analysis will be provided without the patient ID code, but with the blinded randomised treatment code (eg, treatment 1 or treatment 2). The calculated interim p value for performing a safety review of the study (using the 1d98 program) is 0.006 (using a one-sided O’Brien-Fleming boundary). The proportion of participants with an unplanned hospitalisation for asthma will be compared with the expected proportion of 2.0% using the binomial test for proportions. If the observed rate exceeds the expected rate with a p value <0.006, a safety review of the study will be undertaken. The p value calculations use the 1d98 program, an alpha spending function, with alpha nominated as 0.05, evenly distributed analysis times, and O’Brien Fleming boundaries. If the findings of the safety analysis indicate a safety review is necessary, then termination of the trial will be considered.
Sample size re-estimation at the blinded interim analysis point
We plan a blinded re-estimation of the required sample size for the trial at the interim analysis point based on the rate of severe exacerbations in each of the arms of the study, masked as to treatment allocation. In the blinded assessment of rate of severe exacerbations in the two treatment arms, if the higher of these two event rates is less than 0.30 events per year, then the sample size requirements will be larger than currently planned. Should a considerable increase in recruitment be required, the study team will consider whether the increase is reasonably achievable. If the team considers that the increased sample size is not achievable, a blinded sample size estimation using an outcome of ‘asthma exacerbations per patient per year’ will be performed. The team will then consider options such as changing the primary outcome variable from ‘severe asthma exacerbations per patient per year’ to ‘asthma exacerbations per patient per year’. An asthma exacerbation is defined as worsening asthma resulting in unplanned medical review or worsening asthma resulting in the use of systemic corticosteroids for any duration. Any change will be made prior to database lock.
Monitoring
The study will be monitored by a clinical trials monitor based at the Medical Research Institute of New Zealand.
An eCRF will be used to randomise subjects into the study, track dispensed medications and enable data entry for each patient.
Discussion
This study represents the first peer-reviewed and independently funded and sponsored RCT of the use of an ICS/fast-onset LABA inhaler as sole reliever therapy in patients with asthma in whom maintenance ICS and SABA reliever therapy is recommended. This trial, being pragmatic and open label in design, and recruiting a broad population of patients with mild asthma taking SABA with or without ICS, will complement the evidence obtained from three current studies of the as-needed budesonide/formoterol regimen in different populations and with different study designs (the double-blind regulatory studies SYGMA 1 and 2)21 and the pragmatic open-label study, Novel START, which is also investigating the efficacy of this regimen.22
A comparison of the design of the two open-label studies, PRACTICAL and Novel START, is shown in table 3. Both studies have few inclusion and exclusion criteria, and have been designed to increase the external validity of the results and to provide a more accurate estimate of real-world effectiveness than can be obtained from double-blind regulatory SYGMA studies in highly selected populations. PRACTICAL is a New Zealand-based study, and it is recognised that this may limit generalisability to other healthcare environments. Additional information will be obtained from the multicountry Novel START study. A feature of these two studies is that they are both enrolling a broad range of adult patients, including smokers (up to a 20 pack year history), with a doctor diagnosis of asthma, but with no specific lung function or reversibility requirements; the participants thus represent patients treated for asthma in the community.32 In both studies, FeNO and peripheral blood eosinophil count will be collected at baseline, allowing combined analysis of biomarker predictors of risk and treatment response. Key differences are that, prior to entry, participants for PRACTICAL may be using low-moderate-dose ICS, whereas Novel START participants are only using as-needed SABA prior to entry; that the comparison in PRACTICAL is with low-dose ICS plus as-needed SABA, whereas the primary comparison in Novel START is with as-needed SABA alone; and while both studies focus on risk reduction as the main goal of treatment in mild asthma, the primary outcome measure for PRACTICAL is severe exacerbations, whereas the primary outcome for Novel START is a composite exacerbation definition.33 Standardisation of key features of the protocols will enable a combined analysis of individual patient data from the PRACTICAL and Novel START trials. For this analysis, data will be combined for the comparison of the ICS/LABA reliever therapy and ICS maintenance and SABA reliever therapy regimens, with the rate of severe exacerbations per patient per year the primary outcome variable.
Comparison of the PRACTICAL and Novel START trial designs
There are some parallels between the design of PRACTICAL and the double-blind regulatory study SYGMA 2 (NCT02224157), with both studies comparing as-needed budesonide/formoterol with regular low-dose budesonide and as-needed terbutaline, and both having severe exacerbations as the primary outcome. However, the more restrictive eligibility criteria for SYGMA 2 and its requirement for 2–4 weeks of run-in period on SABA only (to confirm the need for step-up treatment) mean that PRACTICAL will provide evidence that is more generalisable to the primary care population.
In PRACTICAL, comparison will be made with maintenance low-dose ICS and SABA reliever therapy, corresponding to the treatment that would be recommended by the GINA strategy report34 for such patients with asthma on their entry into the study. The dose of budesonide in the comparator arm is based on its established dose–response relationship in asthma, in which 400 µg/day achieves around 80%–90% of the maximum obtainable efficacy for all major outcome measures including severe exacerbations.35 In the initiation of ICS therapy budesonide 400 µg/day or equivalent achieves maximum efficacy.36 The dose of budesonide/formoterol 200/6 µg one inhalation as required for symptom relief is one of the doses recommended for reliever therapy use in the Symbicort as Maintenance And Reliever Therapy (SMART) regimen.15 19
While the SYGMA studies are double-blinded regulatory RCTs, in PRACTICAL and NovelSTART the medications will not be administered double-blind. This is because this would mean that two of the potential ‘real world’ advantages of the ICS/LABA reliever therapy regimen, which are the use of a single medication and no requirement for regular inhaler use, would be lost. As such, the PRACTICAL study will have good external validity. Adherence to medication is always greater during a clinical trial, but if participants are required to take multiple dummy inhalers every day their behaviour is likely to be very different from that seen when taking a PRN only medication and that might serve to reduce the difference between the groups. Removing blinding will allow patterns of use to be closer to those seen in usual clinical practice. Likewise participants will not be required to measure their peak flow or to fill in a diary every day as this is not part of normal clinical practice, and could potentially prompt the participants randomised to ICS to take their medicines regularly and promote adherence. The PRACTICAL and NovelSTART studies should therefore produce evidence that is complementary to the SYGMA studies and gives a better estimate of likely effectiveness outside of clinical trials.
Participants randomised in PRACTICAL will also be provided with written asthma action plans, based on standardised plans developed by the Asthma Foundation of New Zealand,37 and the National Asthma Council Australia.38 The purpose of these plans is to reinforce the randomised treatment regimens and provide written instructions on what actions the participants should take in the situation of worsening asthma, in particular when to start prednisone, seek GP review and emergency medical care in the situation of an exacerbation, and the maximum daily doses of budesonide/formoterol and terbutaline for the different regimens, before medical attention should be sought. The action plans will also serve to facilitate standardised assessment and recognition of exacerbations by participants in the clinical trial.
In the electronic monitoring substudy, participants will be told that they are using a modified inhaler that has been produced specifically for this study to count the precise number and timing of doses used during the study period. This will provide a reason for the participants why they need to avoid using other inhalers. Participants will be told that the purpose of the study is to compare the benefits of the treatment regimens and to determine whether the patterns of use influence outcome.
The primary outcome variable is the severe asthma exacerbation rate, expressed as number per patient per year. A severe asthma exacerbation is defined according to the ATS/ERS criteria.27 The assumed rate of severe exacerbations per patient per year of 0.30 for the ICS+SABA group is derived from RCTs that have reported a rate of 0.21 in steroid-naïve subjects treated with budesonide 200 µg/day (using the same criteria for severe exacerbations, peak flow criteria excluded) and rates in subjects previously treated with ICS at baseline of 0.92 and 0.96 (budesonide 200 and 400 μg/day),39 0.35 (budesonide 800 µg/day),40 and 0.35 (budesonide 400 µg/day).41 The conservative proposed reduction in severe exacerbations is based on the reported non-significant 38% reduction with ICS and SABA reliever therapy (given by separate inhalers) when compared with physician-adjusted maintenance ICS and SABA reliever therapy.18 This 38% reduction in severe exacerbations would be expected to be less than that observed in the proposed study, due to their study of highly compliant patients, the use of separate inhalers rather than a combination inhaler and ICS/SABA rather than ICS/LABA reliever therapy.
In current asthma guidelines, ICS/LABA treatment is recommended only as a step-up when asthma is not well controlled by treatment with maintenance low-dose ICS plus SABA reliever. These recommendations are based not on efficacy, but on the higher cost of the combination medications.42 However, if as-needed low-dose ICS with rapid-onset LABA is effective for risk reduction in patients with mild asthma, new cost-effectiveness analyses will be needed, not only with current pricing schedules but also for potential future generic medication options. The cost-effectiveness data collected in PRACTICAL will contribute to these analyses.
In conclusion, this independently funded study will investigate a novel treatment regimen that might contribute to a paradigm shift in asthma management for adults with mild to moderate asthma for whom regular anti-inflammatory treatment is currently recommended, by providing evidence of its effect on exacerbation risk, asthma symptom control, degree of airway inflammation and cost-effectiveness.
Acknowledgments
Some of the content of this paper has been reproduced fromthe Protocol, which was previously made available online through the AustralianNew Zealand Clinical Trials Registry (ANZCTR) at www. anzctr. org. au Trial ID:ACTRN12616000377437.
Acknowledgments
Some of the content of this paper has been reproduced from the Protocol, which was previously made available online through the Australian New Zealand Clinical Trials Registry (ANZCTR) at www.anzctr.org.au Trial ID: ACTRN12616000377437.
References
Footnotes
Twitter @ClinResearchNZ
Contributors RB conceived the study and finalised the study design and secured funding from the HRC and Genentech with the assistance of the steering committee. JP wrote the first draft of the protocol. JF and RB wrote the first draft of the manuscript. All authors were involved in the design of the study protocol and reviewed this manuscript prior to submission.
Funding The primary funding is being provided by the Health Research Council of New Zealand, through a programme grant to the MRINZ: reference number 15/573. The PRACTICAL study has undergone full external peer review as part of the funding process. The funding agreement is made between the HRC and MRINZ, as Sponsor. Funding for the collection materials, shipping and analysis of periostin and future unspecified research samples will be provided by Genentech Inc. This funding agreement is made between Genentech and MRINZ, as Sponsor.
Competing interests JF has received support to attend educational meetings from AstraZeneca and Boehringer Ingelheim. JP is an HRC Clinical Research Training Fellow. AC is a Pharmac PTAC respiratory subcommittee member and has received honoraria from AstraZeneca and GlaxoSmithKline for speaking and advisory board work. HKR has participated in advisory boards for AstraZeneca (including the Steering Committee for the SYGMA studies), GlaxoSmithKline, Merck and Novartis; has presented independent medical education at symposia funded by AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novartis, Mundipharma and Teva; and has received independent research funding from GlaxoSmithKline and AstraZeneca. RB has participated in advisory boards for AstraZeneca, GlaxoSmithKline and Novartis; received research grants from AstraZeneca, Cephalon, Genentech, GlaxoSmithKline, Novartis and Sanofi Aventis; and received payment for lectures or support to attend meetings from AstraZeneca and GlaxoSmithKline. Other authors have no relevant competing interests.
Ethics approval Northern B Health and Disability Ethics Committee of New Zealand.
Provenance and peer review Not commissioned; internally peer reviewed.