Article Text

Abstract
Introduction Malignant pleural effusion (MPE) is common and currently in UK there are an estimated 50 000 new cases of MPE per year. Talc pleurodesis remains one of the most popular methods for fluid control. The value of thoracic ultrasound (TUS) imaging, before and after pleurodesis, in improving the quality and efficacy of care for patients with MPE remains unknown. Additionally, biomarkers of successful pleurodesis including measurement of pleural fluid proteins have not been validated in prospective studies.
The SIMPLE trial is an appropriately powered, multicentre, randomised controlled trial designed to assess ’by the patient bedside' use of TUS imaging and pleural fluid analysis in improving management of MPE.
Methods and analysis 262 participants with a confirmed MPE requiring intervention will be recruited from hospitals in UK and The Netherlands. Participants will be randomised (1:1) to undergo either chest drain insertion followed by instillation of sterile talc, or medical thoracoscopy and simultaneous poudrage. The allocated procedure will be done while the patient is hospitalised, and within 3 days of randomisation. Following hospital discharge, participants will be followed up at 1, 3 and 12 months. The primary outcome measure is the length of hospital stay during initial hospitalisation.
Ethics and dissemination The trial has received ethical approval from the South Central-Oxford C Research Ethics Committee (Reference number 15/SC/0600). The Trial Steering Committee includes an independent chair and members, and a patient representative. The trial results will be published in a peer-reviewed journal and presented at international conferences.
Trial registration number ISRCTN: 16441661.
- pleural disease
- mesothelioma
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Malignant pleural effusion (MPE) is common. Currently in UK there are an estimated 50 000 new cases of MPE each year.1 2 This translates to one new case per 1000 population. The incidence of MPE is increasing in an ageing population—an extra 100 000 cases of cancer are expected per year by 2025.3 The majority of patients with MPE develop disabling symptoms, particularly breathlessness and chest pain.4 Removal of pleural fluid by intercostal drainage alleviates symptoms, but in the majority, MPE recurs after single drainage (recurrence rate ~100% at 1 month).4 To reduce this morbidity, adherence of the pleura (pleurodesis) to prevent fluid accumulation is advocated.4 5
Pleurodesis using sterile talc is the current standard of care for MPE.4 Talc creates a chemical injury to the parietal pleura which provokes inflammation and subsequently adheres the inflamed parietal pleural and visceral lung surface, creating a fibrotic seal between the two surfaces which prevents further fluid accumulation. Talc pleurodesis in randomised studies has a success rate of 70%–80%,5 and is associated with modest equipment cost (~£10 sterile talc, ~£40 chest drain: UK costs, current: June 2017, including value added tax (VAT)). However, talc pleurodesis requires insertion of a chest drain and thus an in-hospital stay while the drain is in situ. The majority of MPE pleurodeses are conducted using the ‘talc slurry’ technique, where a drain is percutaneously inserted into the pleural space, all fluid is drained over a 24–48-hour period and talc is then administered via the drain into the now dry pleural space. Alternatively, talc may be administered using a thoracoscopic ‘poudrage’ technique where the pleural space is fully drained with the aid of endoscopic cameras inserted into the pleural space, and talc applied using pressurised gas during the procedure.
The mean hospital stay following talc slurry pleurodesis is 6 days (SD 4.5) while the mean stay is 4 days (SD 3.5) following poudrage pleurodesis.6 Length hospitalisation is related to the time spent in hospital after talc application and with the chest drain in situ. Using current data on the cost of hospital beds/day6 in the UK (June 2017: including VAT), a 6-day stay for talc pleurodesis is associated with a cost of £1320 in bed day/patient. The overall hospital costs associated with talc pleurodesis are substantial. Assuming conservatively that 50% requires pleurodesis, this equates to 25 000 pleurodeses/year, and with an average hospital stay/patient of 6 days, the UK cost in bed days of pleurodesis is £33 million/year.7
Several attempts have been made to address the optimal timing of talc administration after chest drain insertion, including amount of pleural fluid drained per day and radiographic appearances. A previous study suggests that the optimal timing of talc administration should be determined by the appearance of the chest radiograph.8 However, there is a paucity of data that addresses when the chest drain should be removed after talc administration, with current UK national guidelines4 citing only two articles providing expert opinion8 9 and two small randomised trials.10 11 Thus, there are no reliable data to inform the clinician of the optimal timing of drain withdrawal following talc pleurodesis.
Use of thoracic ultrasound (TUS) for detection of pleural fluid and to guide pleural interventions is becoming widely available and pleural adherence may be detectable via TUS.12 An observational study has shown that fibrin strands on ultrasound may be associated with higher pleurodesis success,13 inferring that sonographic markers may provide prognostic information. Sonographic evidence of pleural adherence using a lack of the normal ‘lung sliding’ sign seen when normal visceral lung moves beneath the parietal pleura predicted pleurodesis success in a rabbit animal model of pleurodesis,13 in which absence of lung gliding (pleural adherence) at 14 days postpleurodesis correlated with histological pleural fibrosis score at animal sacrifice. A study (n=10) in humans using the same technique has shown promising results,14 in assessing the success of surgical pleurodesis in spontaneous pneumothorax at 4 weeks. This observation suggests that a TUS scanning protocol for pleurodesis is feasible in humans. However, no previous studies have been made to prospectively assess a TUS pleurodesis protocol in MPE in order to investigate the timing of scanning, or efficacy of the suggested protocol.
In this trial, bedside use of TUS diagnosis of pleural adherence within 24–48 hours of talc administration is the novel application proposed, with the primary outcome being reduction of hospital stay. Pilot data from our department assessing 10 patients with MPE suggest pleurodesis may occur within 24 hours of talc administration and can be detectable using TUS. Additionally, combination of clinical, radiological and biological variables will be done in order to prospectively develop a predictive score for pleurodesis success and survival in patients with MPE.
Methods and analysis
SIMPLE is a multicentre randomised controlled trial designed to evaluate whether use of TUS, in hospitalised patients with MPE before and during the first 24–72 hours post-talc administration, accurately identifies pleural adherence early in treatment, permitting shorter hospital stay without adversely affecting pleurodesis success. The trial (unblinded and postrandomisation) will also interrogate pleural fluid protein expression profiles to determine factors that permit prediction of pleurodesis success and survival. The trial is sponsored by the University of Oxford and coordinated by the Oxford Respiratory Trials Unit (ORTU), University of Oxford. The trial is registered on the International Standardised Randomised Controlled Trial Registry (ISRCTN16441661) and funded by the Marie Curie Institute. The trial is included in the NIHR Clinical Research Network portfolio (ID: 20343). The trial will be conducted in accordance with the Declaration of Helsinki and Good Clinical Practice.
Participants will be randomised to standard care or TUS guided care.
Standard care: Participants will have either thoracoscopy with talc poudrage or a chest drain with talc slurry. Talc slurry pleurodesis will occur as indicated by radiological results (chest X-ray (CXR) after drainage). The drain will be removed postpleurodesis, at 24 hours or later, provided that fluid drainage is less than 250 mL in 24 hours, the lung remains fully re-expanded and there is satisfactory evacuation of pleural fluid on the CXR as per usual practice and guideline-based management.
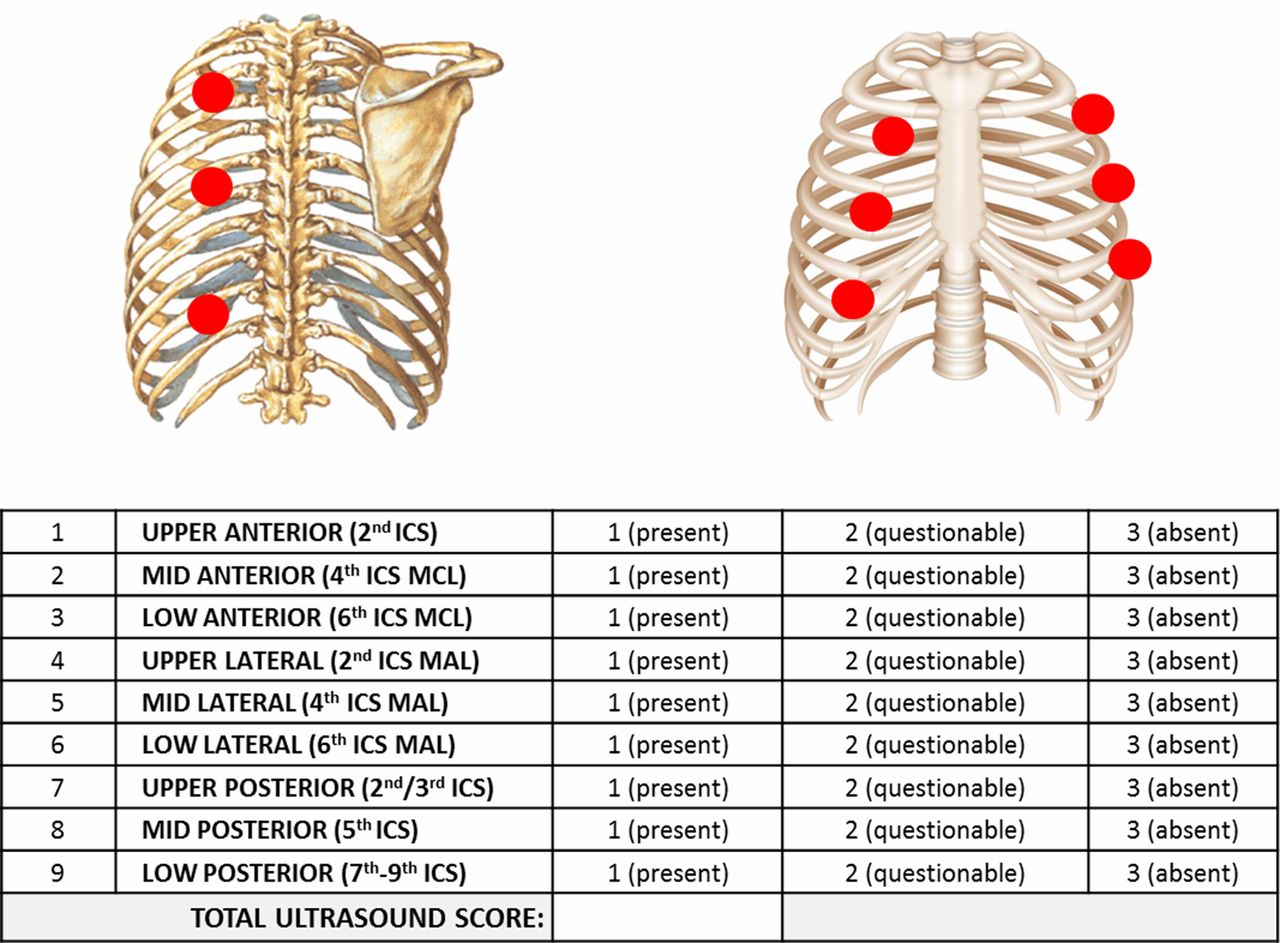
TUS guided care: Participants will have either thoracoscopy with talc poudrage or a chest drain with talc slurry. The pleurodesis will be performed once there is ultrasound scan evidence of effusion resolution. Previous research studies within ORTU have developed a TUS score by assessing lung sliding in nine different sites (anteriorly, posteriorly and laterally) and score with 1 or 2 or 3 if lung sliding is present, questionable or absent (figure 1). The drain will be removed postpleurodesis on the basis of bedside ultrasound appearances: either the sonographic pleural adherence score of >20 or complete pleural adherence in the midaxillary line region.
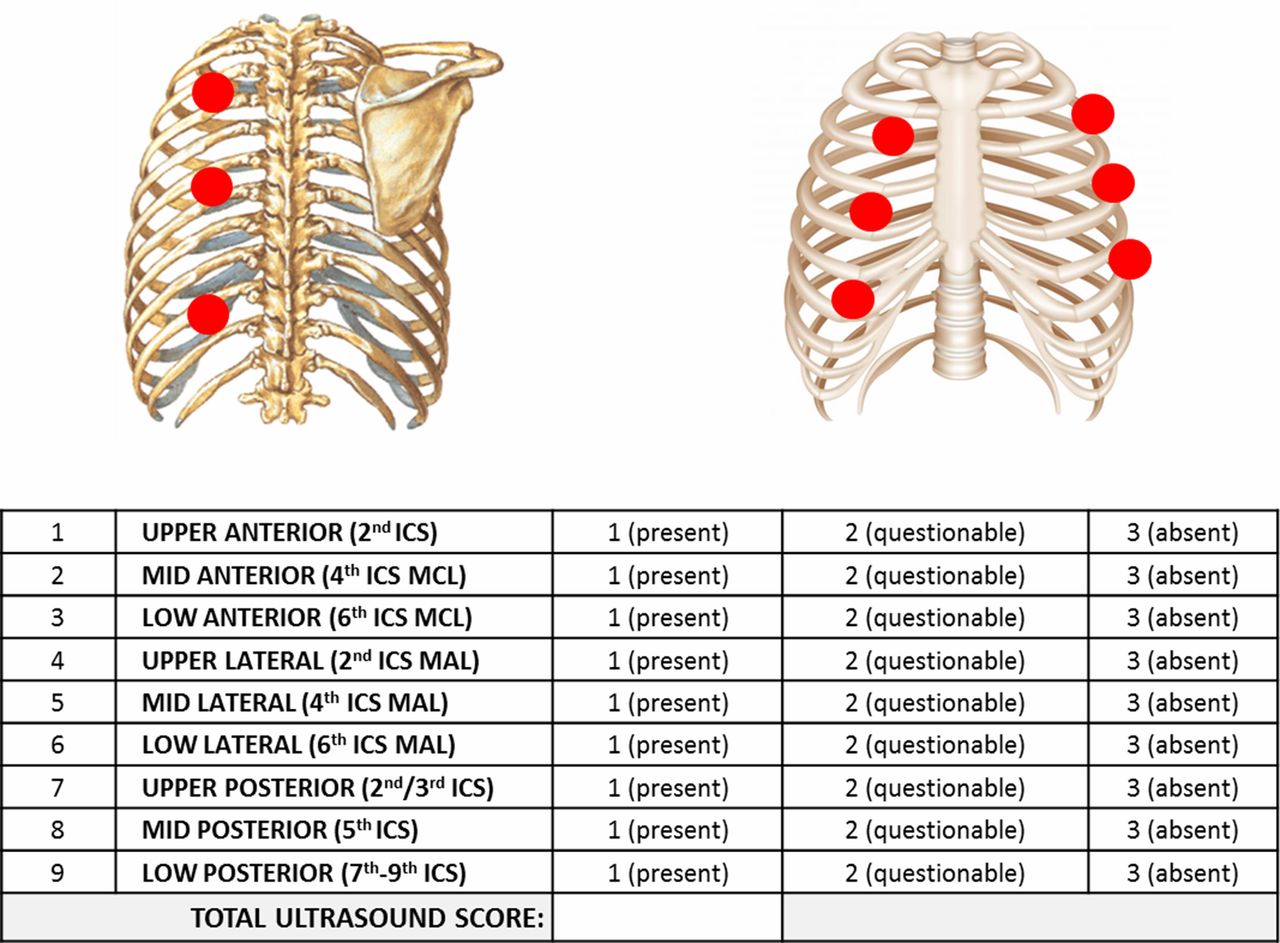
Ultrasound scanning protocol used for SIMPLE study. ICS, intercostal space; MAL, midaxillary line; MCL, mid-clavicular line.
All other interventions will be identical in the treatment arms, including access to further pleural procedures in the case of pleurodesis failure, cancer-specific treatments and supportive care including palliative measures.
The primary research question for participants with a confirmed MPE treated with talc pleurodesis (either slurry or poudrage) is: Does TUS guided care decrease the number of days spent in hospital during the initial hospitalisation when compared with standard management (CXR and fluid output)?
The secondary research questions are:
Can the pleural fluid protein profile predict pleurodesis success at 1 and 3 months postrandomisation?
Does TUS guided care reduce the time of chest drain in situ postpleurodesis when compared with talc slurry?
Does management with TUS guided care cause less breathlessness and thoracic pain for the first 4 weeks postrandomisation when compared with standard care?
Does TUS guided care improve health-related quality of life over 3 months postrandomisation when compared with standard care?
Is TUS guided care cost-effective and reduces healthcare utilisation over 3 months when compared with standard care?
Is TUS guided care reducing time to pleurodesis failure within the first 3 months compared with standard care?
Are there any biological, clinical and radiological biomarkers to predict mortality at 12 months?
Setting
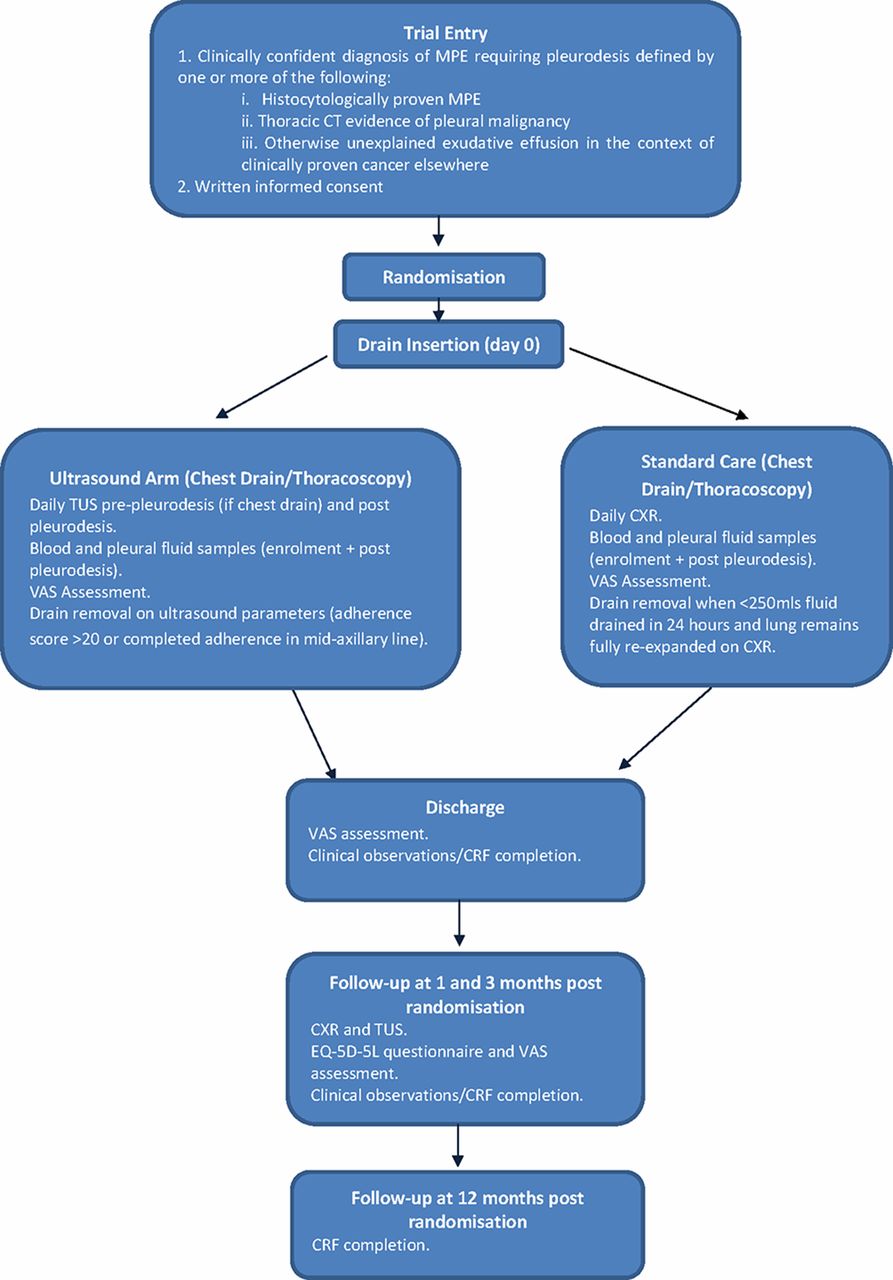
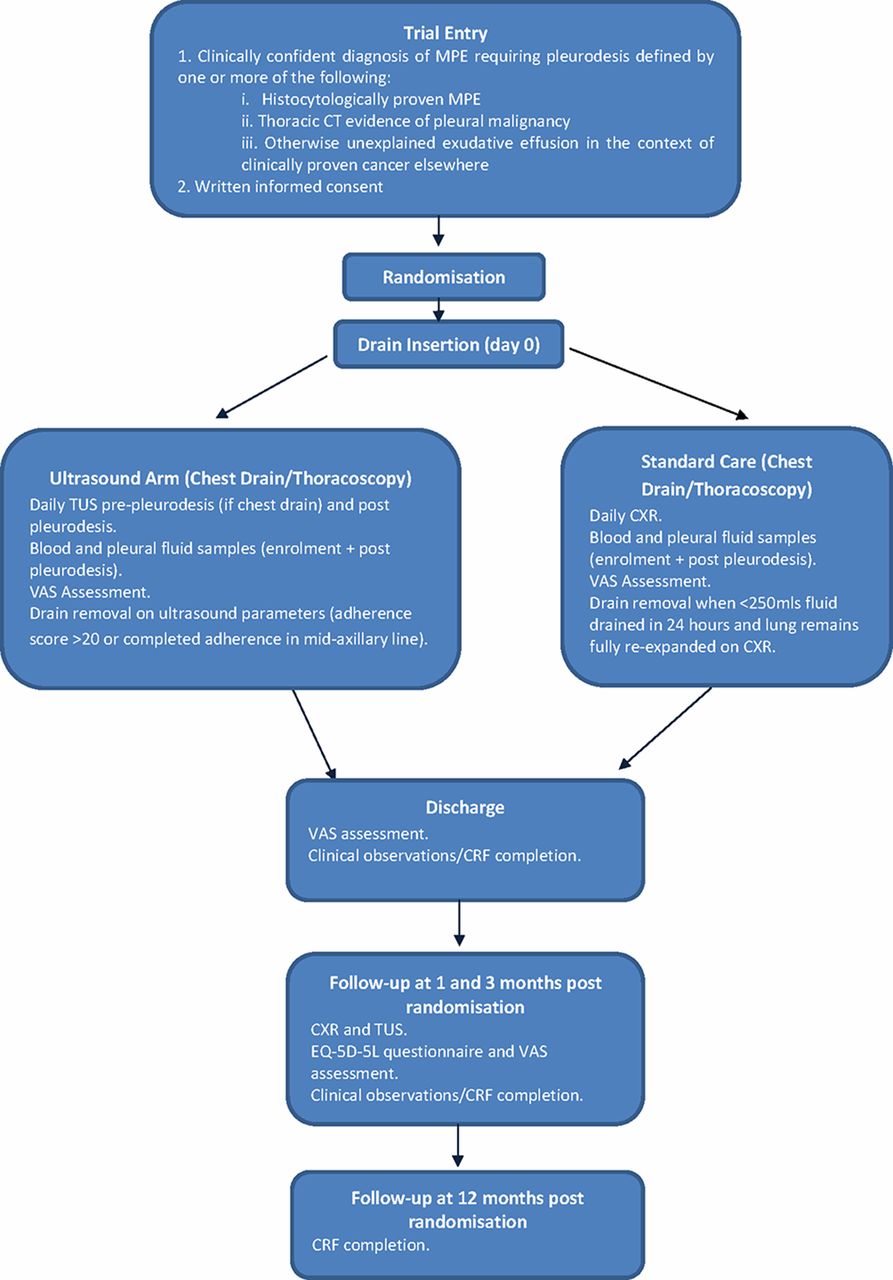
Two hundred and sixty-two participants requiring a pleurodesis intervention for a confirmed MPE will be recruited from UK and Netherlands hospitals. Participants undergoing talc pleurodesis (either slurry or poudrage) will be randomised to undergo either standard care (according to British Thoracic Society (BTS) guidelines) or assessment with TUS. The trial flow diaphragm is shown in figure 2.
{kind=link}
{kind=link}
Study algorithm for SIMPLE study. CRF, clinical report form; CXR, chest X-ray; EQ-5D-5L, EuroQoL 5 Dimensions 5 Levels; MPE, malignant pleural effusion; TUS, thoracic ultrasound; VAS, Visual Assessment Scale.
Subject screening and selection
Participants with MPE will be identified following early discussion at each centre’s cancer multidisciplinary team meetings, at routine outpatient appointments and during inpatient reviews. Eligible participants will be invited to participate and will be provided with a participant information leaflet at the earliest opportunity. Participants can be enrolled only once into the SIMPLE trial.
Inclusion criteria
Clinically confident diagnosis of MPE requiring pleurodesis defined as any of the following (more than one can be included):
Histocytologically proven MPE.
Thoracic CT evidence of pleural malignancy.
Otherwise unexplained exudative effusion in the context of clinically proven cancer elsewhere.
Written informed consent.
Exclusion criteria
Age <18 years old.
Poor prognosis (defined as patients in whom pleurodesis will not be offered in normal practice).
Irreversible contraindication to drain insertion.
Informed consent
Written and verbal versions of the Participant Information Sheet and Consent form will be presented to the participants detailing the exact nature of the trial; what it will involve for the participant; the implications and constraints of the protocol; and the known side effects and any risks involved in taking part. It will be clearly stated that the participant is free to withdraw from the trial at any time for any reason, without prejudice to future care, and with no obligation to give the reason for withdrawal.
Participants will be given sufficient time (in their own opinion) to fully consider trial entry, as well as to ask questions of investigators. The consent form (see online supplementary appendix 3) will be countersigned by either a medical or nursing member of the trial team.
Randomisation
Once the participant has given written consent to the trial, a member of the trial team will complete a randomisation form and randomise the participant to a treatment allocation and trial number. Randomisation should occur after the participants undergo their baseline assessment.
The randomisation process will be 1:1 and performed by sites using a web-based randomisation system. Allocation to interventional or control arm will be performed by an authorised and trained member of the site team who will enter the eligibility criteria data and minimisation factors onto a computer to confirm eligibility and derive the allocated treatment. The randomisation system will be provided by Sealed Envelope Randomisation Services (Sealed Envelope, Concorde House, Grenville Place, London). Minimisation with a residual randomised component will occur for the minimisation factors of:
Centre.
LENT score (categories: low score 0–1, moderate score 2–4, or high risk score 5–7); it is a scoring system that predicts survival in people with MPE.15
Intention to treat (ITT) with talc poudrage or talc slurry.
Participants and clinicians will not be blinded to treatment allocation.
Intervention arm
As part of the trial, all participants randomised to the intervention arm will have TUS assessments. For participants having a chest drain with talc slurry, these will occur prepleurodesis and postpleurodesis (for up to 3 days or until declared fit for discharge). For participants having a thoracoscopy with talc poudrage, these will only occur postpleurodesis (for up to 3 days or until declared fit for discharge) as there is no pretalc period of drainage in thoracoscopies.
All daily TUS assessments should be recorded on trial clinical report forms (CRF). The information required will include the size of any residual effusion (based on the number of rib spaces where pleural fluid is visible and the maximum depth of the effusion measured in centimetres) and characteristics of this residual fluid including echogenicity and presence of septations. Additional measurements will be taken using B-mode ultrasound specifically assessing and scoring the movement or ‘sliding’ of the underlying lung in nine specific regions (upper, middle and lower zones in the anterior (mid-clavicular line), lateral (midaxillary line) and posterior chest). Lung sliding will be scored as either present (=1), questionable (=2) or absent (=3) in nine regions, consistent with previous animal studies in this area of practice. This will generate a total pleural adherence score for the hemithorax being assessed.
TUS imaging will be essential in defining both the time to perform pleurodesis and also the removal of the chest drain. Specifically, pleurodesis with talc slurry will be performed once there is a small effusion on TUS (less than two rib spaces) or the maximum depth of the fluid will be less than 2 cm in two out of three basal areas (posterior/midaxillary line/anterior) on TUS. Pleurodesis will be performed as per normal clinical practice proposed by the current BTS guidelines (4 g of talc in 50 mL normal saline—0.9% with intrapleural local anaesthesia). The drain will be removed postprocedure on the basis of bedside ultrasound appearances: either mean sonographic pleural adherence score of >20 or complete pleural adherence in the midaxillary line region. If after 72 hours postpleurodesis there is minimal fluid drainage, good CXR appearance but the lung is sliding on TUS, the participant will be managed as per BTS guidelines (removal of the chest drain) and this will be documented on the CRF.
Ultrasound still images and video clips (5–10 s in duration) will be recorded in order to allow verification of the findings, after trial completion, by an independent assessor who will be blinded to the participant’s clinical status; this assessor will, as a minimum standard, hold level 1 Royal College of Radiologists accreditation in thoracic ultrasound. All images and video clips will be anonymised for the trial participants and copied to encrypted external hard drives. To ensure no loss of data, images will be transferred at two time points in the trial; discharge and after the 3-month follow-up. All encrypted external hard drives will be couriered to ORTU to be securely stored on the Trust server and then analysed by the independent assessor at the end of the trial.
Control (standard care) arm
For all participants during initial drainage, a daily CXR will be conducted. In the control arm group (standard care), the talc slurry pleurodesis will be administered when the patient meets standard criteria (suitable apposition of lung and chest wall on CXR, less than 250 mL pleural fluid drainage per day as per current BTS guidelines) and will be conducted according to the trial-specific instructions on talc.
Once talc pleurodesis has been conducted, daily observations will be recorded on the CRFs from the clinical notes. The drain will be removed when the participant meets the objective drain removal criteria, based on current BTS guidelines, and will be recorded on CRFs. For participants treated with talc poudrage via thoracoscopy, the chest drain will be removed based on the same criteria in the guidelines.
TUS may be used in the non-ultrasound arm if clinically indicated for specific reasons (eg, poor drain output, to assess drain position) as per usual clinical care. The clinician will be required to clearly document the clinical reason(s) for using TUS in the control arm on a CRF.
Trapped lung
For patients in both arms, treated with either talc slurry or poudrage, if there is evidence of trapped lung postchest drain insertion (defined on CXR as lack of pleural apposition in more than two-thirds judged by local investigator), the participants can still remain in the trial but they should be managed as per BTS guidelines. It will be up to the discretion of the treating physician as to whether they still perform pleurodesis.
If the participant is in the ultrasound arm, the TUS assessment is not feasible and the management of the participant should not be planned according to TUS characteristics. Participants with trapped lung in both arms will be followed up as per protocol.
Visual Assessment Scale scoring
All participants should document a Visual Assessment Scale (VAS) score for both thoracic pain and breathlessness during their baseline assessment. The participant should then complete daily scores for 7 days postpleurodesis, then weekly until the 1-month follow-up appointment.
Participant diaries
Participants will be provided with preprinted diaries to keep with them during their involvement in the trial. The diaries will capture all personal contacts that the participant may have had with healthcare services outside of the trial. These diaries will be used at follow-up appointments, in addition to medical notes, to determine the health utilisation of each participant during the follow-up period.
Biological samples and storage
All participants consenting to trial sample collection should have the following blood (10 mL) and pleural fluid (20 mL) samples taken at certain stages of the trial:
Enrolment.
Postpleurodesis (maximum 24 hours postpleurodesis).
All trial samples should be posted securely to ORTU and will be analysed with specific laboratory-based techniques to identify blood and pleural fluid biomarkers that will predict pleurodesis success and patients’ favourable survival. Pleural fluid and blood samples postpleurodesis will be tested to define the biological effects of talc pleurodesis in both arms. Samples will be centrifuged in the ORTU and stored under the Human Tissue Agency licence for future use subject to ethical approval.
Data management
CRFs will be completed by the trial team at recruiting centres and sent to the ORTU. Data will then be entered onto the trial database (OpenClinica clinical trials software). Missing data and data queries will be highlighted to the trial teams on a monthly basis. The CRFs will only identify participants using their personal trial identification number (no identifiable participant information).
Discontinuation/withdrawal of participants from trial
Participants have the right to withdraw from the trial at any time without having to give a reason and this will not affect their future care. Withdrawal details should be recorded on the withdrawal/loss to follow-up CRF. In addition, the investigator may discontinue a participant from the trial at any time if the investigator considers it necessary. Withdrawals from the trial should be discussed with the trial clinical coordinator, the chief investigator or their deputy.
Loss to follow-up will be minimised by diligent liaison with the patient, their oncology team and general practitioner. Any loss to follow-up will be recorded on the withdrawal/loss to follow-up form.
Primary outcome
The primary outcome of the trial is the length of hospital stay (in days) during the initial hospitalisation.
Secondary outcomes
The trial’s secondary outcomes are the following:
Pleurodesis success at 1 and 3 months postrandomisation.
Number of days in hospital postrandomisation with drain in situ.
EuroQoL 5 Dimensions 5 Levels questionnaire—at enrolment, 1 month and 3 months postrandomisation.
Patient-reported dyspnoea/chest pain (VAS assessment) at baseline and postpleurodesis (daily for 7 days and then weekly for 4 weeks).
Healthcare utilisation logs at discharge, 1 month and 3 months.
Mortality at 12 months.
Pleurodesis failure at 1 and 3 months postrandomisation.
Sample size calculations
A minimum total sample size of 262 participants will be recruited to this trial.
Hospital stay: superiority comparison. Mean hospital stay is 6 days (SD 4.5) for talc pleurodesis with ‘standard’ care. Assuming (conservatively) that ultrasound reduces stay by 2 days, 254 patients randomised 1:1 required to demonstrate this difference (90% power, 5% two-sided significance), which includes 5% attrition due to death before discharge. This difference includes the clinically significant difference to detect a 2-day difference in hospital stay.
Pleurodesis success: non-inferiority comparison. Results from well-conducted randomised trials demonstrate 70%–80% pleurodesis efficacy at 3 months using talc as the pleurodesis agent. Assuming mean efficacy to be 75%, and a non-inferiority margin (delta) of 15%, 262 patients are required to demonstrate non-inferiority (80% power, 2.5% one-sided significance).
Statistical analysis plan
A summary of the planned statistical analysis is included here. The primary analysis for each outcome will be by ITT. If a continuous variable is normally distributed, we will use t-tests (or analysis of covariance) to compare the intervention at the optimal dose with the control group, and will present the results by mean difference and the corresponding 95% CI. If a continuous variable is not normally distributed, a non-parametric test will be used for the analysis. For categorical variables including quality of life, χ2 tests will be used for comparing treatment groups. P values will be reported to three decimal places. All tests will be considered statistically significant at the 5% level and all comparative results will be presented as summary statistics with 95% CIs and reported according to the appropriate Consolidated Standards of Reporting Trials extension. All analyses will adjust for minimisation variables and these will be included as covariates in the regression model for each outcome.
Specific description of statistical methods:
Hospital stay: This will be measured from randomisation until discharge using objective criteria. The analysis will be undertaken on the ITT population. Sensitivity analyses will be undertaken on the per-protocol population.
Pleurodesis success: This will be measured using blinded assessment and objective criteria (at 1 month and 3 months). Non-inferiority will be assessed against the lower limit of the CI to determine whether it does not cross the prespecified non-inferiority margin. The non-inferiority comparison for pleurodesis success will be based on per-protocol analysis, which is more conservative than the ITT.
Modelling of predictive factors: At trial completion, analysis of potential predictive factors for successful pleurodesis and survival will occur using standard statistical methods, examining the relationship between outcome (pleurodesis success, patient-reported outcome measures) and factors individually first, then combining into a regression model to assess their independent effects.
Health economic analysis: An economic evaluation adherent to guidelines for good economic evaluation practice will be undertaken integral to the main trial. A within-trial cost-effectiveness (additional cost per pleurodesis success achieved) and cost-utility analysis (additional cost per quality-adjusted life years gained) will explore the 3-month cost-effectiveness of TUS guided care when compared with standard care, with incremental cost-effectiveness ratios being calculated by dividing the difference in healthcare costs by the difference in effects.
End of trial
Trial closure will be when all final sample analyses have been completed or at the discretion of the Trial Steering Committee (TSC) acting on the recommendation of the Independent Data Monitoring Committee (IDMC).
Ethics and dissemination
Monitoring
An IDMC will be convened at regular intervals, consisting of members who are independent of the trial investigators. The role of the IDMC is to monitor the main outcome measures, data quality and completeness and provide advice to the TSC, specifically as to whether recruitment can continue.
Safety reporting
No significant safety concerns are anticipated in relation to any additional measurements or procedures carried out specifically as part of this trial. The proposed trial intervention (TUS) is non-invasive and not expected to present any additional risk to participants or result in any complications. Given the low-risk nature of the trial intervention, no specific safety reporting is felt to be required.
Trial monitoring and oversight
The TSC will be responsible for overseeing the progress of the trial and will meet at least once a year. The TSC will comprise independent chairperson, independent members, statistician, patient representative and the chief investigator.
Dissemination
The trial will be publicised at regional and national conferences. The final results will be presented at scientific meetings and published in a peer-reviewed journal (authorship will be according to the journal’s guidelines). In addition, a lay summary of the trial results will be circulated to potentially interested parties.
References
Footnotes
Contributors IP and NMR conceived the initial trial concept. All authors (IP, HEGP, AY, NIK, GK, SM, LC, JPC, NR, MD, RFM, NMR) contributed to the development of the trial design and protocol. NMR, SM and LC carried out the sample size calculations. SM, LC, IP and NMR wrote the statistical analysis plan. All authors have read and approved this manuscript.
Funding IP is the recipient of a REPSIRE2 European Respiratory Society Fellowship (RESPIRE2-2015-7160). NMR is funded by the National Institute Health Research (NIHR) Oxford Biomedical Research Centre. SIMPLE study is funded by the Marie Curie Institute (C49481/A17178).
Competing interests None declared.
Ethics approval South Central-Oxford C Research Ethics Committee (Reference number 15/SC/0600).
Provenance and peer review Not commissioned; externally peer reviewed.