Article Text

Abstract
Background Treatment of infective bronchitis involving Pseudomonas aeruginosa is a cornerstone of care in patients with cystic fibrosis (CF). This phase IIb, randomised, double-blind, placebo-controlled study assessed the efficacy and safety of ciprofloxacin dry powder for inhalation (DPI) in this population.
Methods Patients with CF, ≥12 years of age (N=286), were randomised to ciprofloxacin DPI (32.5 mg (n=93) or 48.75 mg (n=93)), or corresponding placebo (32.5 mg, n=65; 48.75 mg, n=35) twice daily for 28 days. The primary objective was the change in forced expiratory volume in 1 s (FEV1) from baseline (day 0) to end of treatment (day 29) in the intent-to-treat population for ciprofloxacin DPI compared with the corresponding placebo group.
Results The primary effectiveness objective was not met; there were no significant differences in change in FEV1 between ciprofloxacin DPI and the corresponding placebo group for either dose (p=0.154). However, in pooled analyses, FEV1 decline from baseline to treatment end was significantly lower with ciprofloxacin DPI than with placebo (pooled data; p=0.02). Ciprofloxacin DPI showed positive effects on sputum bacterial load and quality of life, but these effects were not maintained at the 4-week follow-up. Ciprofloxacin DPI was well tolerated and there were no significant differences in type/incidence of treatment-emergent adverse events by treatment group (p=0.115).
Conclusions Further investigations are needed to determine the full scope of the beneficial effects of ciprofloxacin DPI for patients with CF.
Trial registration number Clinicaltrials.gov NCT00645788; EudraCT 2008-008314-40.
- Cystic Fibrosis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
In this placebo-controlled, randomised, phase IIb clinical trial, 288 adolescent and adult patients with cystic fibrosis were assigned to receive placebo or ciprofloxacin dry powder for inhalation (DPI) (doses of 32.5 mg or 48.75 mg) twice daily for 29 days.
There were no significant differences in the primary efficacy end point of change in forced expiratory volume in 1 s (FEV1) between ciprofloxacin DPI and the corresponding placebo group for either dose, but in pooled analyses, ciprofloxacin DPI was associated with a significantly lower decrease in FEV1 compared with placebo.
Although this study did not achieve its primary efficacy end point, ciprofloxacin DPI showed signs of efficacy, and was safe and well tolerated during treatment.
Introduction
Cystic fibrosis (CF) is a chronic, incurable and life-shortening disease, with respiratory failure resulting from chronic pulmonary infection accounting for the majority of deaths.1 ,2 Treatment of chronic respiratory infection is therefore a cornerstone of CF care.1 Of the organisms responsible for pulmonary infection in CF, Pseudomonas aeruginosa is one of the most prevalent,1 with an estimated 50.6% of patients chronically infected with this pathogenic bacteria in the USA in 2011.3 Management of chronic infection consists of airway clearance using techniques such as postural drainage, chest compression, oscillatory positive expiratory pressure or exercise,4 combined with antibiotic treatment. Antibiotics may be administered orally, via an intravenous line or via inhalation.5
The most frequently used US Food and Drug Administration (FDA)-approved inhaled antibiotics require the use of compressors and nebulisers, and may require long treatment durations.6 Furthermore, nebulisers need to be carefully cleaned after use, and these treatments are relatively expensive to employ. All these factors are barriers to the effective use of this mode of administration.6 ,7 Dry powder inhalers, which are small, portable, breath-actuated devices,8 have proven useful in the treatment of asthma and other respiratory conditions.6 Importantly, they can be administered within a short period of time, thereby helping to reduce the burden of treatment. This is an essential factor to consider in a patient population that spends an average of 2 h daily on healthcare-related activities.9 ,10 An important aspect of the CF therapeutic drug development pipeline has been to develop a wider range of agents and delivery systems to treat P. aeruginosa respiratory infection,11 such as the recently approved aminoglycoside antibiotic tobramycin DPI (TOBI Podhaler).12 Despite this approval, there is still a clinical need for antibiotics that can be administered via the DPI route for the treatment of P. aeruginosa in patients with CF.6 ,11 ,13
Ciprofloxacin DPI is a fluoroquinolone antibiotic manufactured using PulmoSphere technology from Novartis (Novartis Pharma AG, Basel, Switzerland) and consists of particles of a controlled size of ≤5 µm, with powder density and morphology optimised for pulmonary delivery.14 ,15 The drug is administered using a capsule-based dry powder inhaler device (T-326 inhaler).16 This formulation of the antibiotic has properties that lead to markedly reduced systemic absorption, thus enhancing its action and potentially minimising side effects.13 ,16–18 After decades of intensive antibiotic use, there is now accelerated evolution of antibiotic resistance. New treatment options administered by inhalation could help counter the increasing drug resistance and improve efficacy.11 ,19 Approval of this agent would provide CF physicians and patients with a new option for antipseudomonal treatments administered through the DPI route.
The mechanism of action of ciprofloxacin is distinct from other agents commonly administered by inhalation, such as tobramycin.20 Ciprofloxacin has frequently been used via oral or intravenous administration for the treatment of pulmonary infection in CF.21 ,22 Therefore, a clinical trial of the new DPI formulation was considered appropriate to perform. A double-blind, prospective, randomised phase IIb trial was designed to assess the efficacy and safety of ciprofloxacin DPI in patients with CF, over a 4-week period.
Methods
Study design
This multicentre, phase IIb, randomised, double-blind, parallel-group, placebo-controlled study (Clinicaltrials.gov trial registration number: NCT00645788; EudraCT number: 2008-008314-40) was conducted at 73 centres: USA (51), Canada (3), Germany (4), Sweden (3), Denmark (1), Israel (4), UK (1) and Australia (6). All study personnel and participants were blinded with respect to active treatment (ciprofloxacin DPI, formulated by Novartis’ PulmoSphere technology, or placebo), but not to dose. The study protocol was amended several times after enrolment started; the most important changes were the inclusion of children aged 12–17 years on availability of safety and pharmacokinetic data, and the addition of a higher ciprofloxacin DPI dose group (48.75 mg) approximately 1 year after the first participants were enrolled. The decision to add the higher dose was based on the good safety results with the 32.5 mg dose and the desire to investigate more than one dose in the phase II trial. Investigation of the higher dose was also recommended by regulatory authorities to help to determine the most appropriate doses to carry forward into the phase III programme.
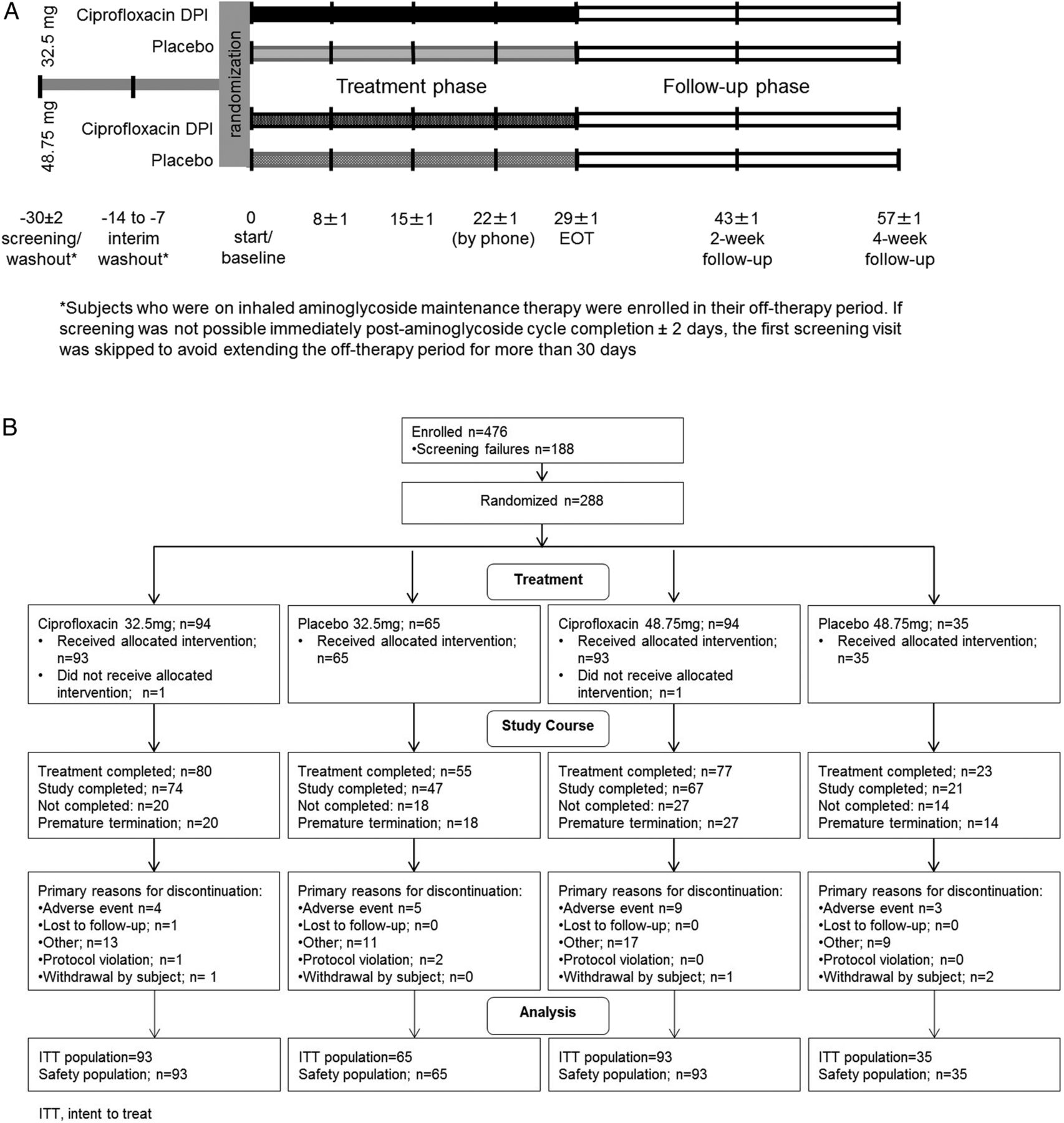
Participants were stratified according to maintenance macrolide usage (ie, participants currently treated with macrolide vs participants not currently treated with macrolide), and randomised 2:2:2:1 to 32.5 mg ciprofloxacin DPI (corresponding to 50 mg dry powder); placebo corresponding to 32.5 mg ciprofloxacin DPI; 48.75 mg ciprofloxacin DPI (corresponding to 75 mg dry powder); or placebo corresponding to 48.75 mg ciprofloxacin DPI (figure 1A). All treatments were given twice daily for 29 days (±1 day). Global Biostatistics, Bayer Pharma AG, assigned the randomisation and drug pack numbers using a central, secure randomisation system. These assignments were then communicated via an interactive voice–response system. All patients maintained their standard treatment, which may have included Pulmozyme (dornase α), albuterol, inhaled steroids, macrolides, saline inhalation and β-agonists (long-acting and short-acting) (stable regimen ≥30 days prior to study drug administration).

Study design (A) and subject disposition (B). (A) Subjects were stratified based on maintenance macrolide usage and randomised 2:2:2:1 to receive twice daily: 32.5 mg ciprofloxacin DPI (corresponding to 50 mg dry powder); placebo corresponding to 32.5 mg ciprofloxacin DPI; 48.75 mg ciprofloxacin DPI (corresponding to 75 mg dry powder); or placebo corresponding to 48.75 mg ciprofloxacin DPI for 29 days (±1 day). DPI, dry powder for inhalation; EOT, end of treatment. (B) A total of 288 patients were randomised. Of these, 286 received either study drug or placebo, and were included in both the ITT and safety analyses sets. ITT, intent-to-treat.
Eligibility
Participants with a documented diagnosis of CF and chronic colonisation with P. aeruginosa, who were in a stable clinical condition and on a stable treatment regimen, were eligible for inclusion in the trial. The main inclusion criteria were age ≥12 years and CF diagnosed by clinical findings and one of the following: a positive sweat chloride test; homozygosity for ΔF508 genetic mutation or a compound heterogeneous genotype for two known CF mutations; a positive sputum or throat swab culture for P. aeruginosa within the previous 12 months. Participants were also required to meet specific entry criteria at the screening visit, specifically: a stable pulmonary status with forced expiratory volume in 1 s (FEV1) ≥35% to ≤75%; room air oximetry ≥88% saturation; absence of antibiotic treatment (with the exception of macrolides, which were allowed concomitantly as part of the standard treatment regimen) for pulmonary exacerbation within 30 days before study drug administration (non-antipseudomonal antibiotics were allowed for other indications); or a stable treatment regimen for 30 days before and throughout the study duration. Patients on long-term on–off cycles of tobramycin or colistin treatment for CF were enrolled in their month off, and visits were scheduled to ensure that the start of study treatment was not delayed beyond the minimum 30-day washout period.
The main exclusion criteria included: findings on screening history and physical examination unrelated to CF that could affect efficacy measurements; colonisation with P. aeruginosa and a ciprofloxacin minimum inhibitory concentration (MIC) of ≥256 µg/mL; respiratory tract colonisation with Burkholderia cepacia complex within the preceding 12 months; symptomatic aspergillosis; haemoptysis ≥300 mL or requiring blood transfusion within the preceding 4 weeks; a history of severe allergies; pregnancy; and participants <18 years with chronic musculoskeletal disease or abnormal musculoskeletal evaluation at baseline, a high risk of chronic or recurrent arthritis or tendonitis, or a history of quinolone-related arthropathy.
All participants (or legal representatives) gave written informed consent and the study was conducted in accordance with the Declaration of Helsinki, and with local legal and regulatory requirements.
End points
The primary objective was comparison of the change in FEV1 from baseline (day 0) to end of treatment (EOT; day 29) between participants with CF randomised to either ciprofloxacin DPI or corresponding placebo in the intent-to-treat (ITT) population (defined as all randomised patients who had received at least one dose of the study medication and had at least one observation after drug administration).
Secondary respiratory objectives included: comparing the change in FEV1 from baseline to days 8 and 15 (treatment phase) and at the 2-week and 4-week follow-up visits; assessing the change from baseline in forced vital capacity (FVC) or maximum forced expiratory flow (FEF25–75%) rate at all subsequent clinic visits; and time to first pulmonary exacerbation requiring any antipseudomonal intervention or hospitalisation. Pulmonary function tests (FEV1, FVC and FEF25–75%) were performed at every clinic visit or at discontinuation. Microbiology on sputum samples obtained at all study visits was performed at a central laboratory. Objectives were assessment of the change in P. aeruginosa density in sputum from baseline to all subsequent clinic visits and the incidence of ciprofloxacin-resistant P. aeruginosa isolates in sputum. Patients completed the CF Quality of Life Questionnaire Revised (CFQ-R; a validated disease-specific instrument that measures health-related quality of life for adolescents and adults with CF) at baseline, EOT and at the 4-week follow-up visit.
Safety
Assessment of the safety profiles for ciprofloxacin DPI and corresponding placebo was a secondary objective of the study. The safety analysis comprised monitoring for adverse events (AEs), and haematology, clinical chemistry, urinalysis, pulse oximetry, vital signs, physical examinations and musculoskeletal examination for patients ≤18 years of age. These were performed at all clinic visits (except for haematology and clinical chemistry at day 15 (treatment phase) and 2-week follow-up visits). AEs were documented as treatment-emergent events (TEAEs) up to 7 days after EOT and, along with serious AEs occurring at any time, were followed up to 30 days post-therapy. Bronchospasm was defined as a drop of ≥15% in FEV1 and pulmonary exacerbations were reported as AEs. Patients who received an antipseudomonal antibiotic for a pulmonary exacerbation were discontinued from the trial.
Statistical analyses
The primary efficacy analysis was based on the ITT population. The randomisation schedule provided a power of >95% to detect a 0.15 L absolute difference (assumed SD: 0.2 L) in change in FEV1 from baseline to EOT for 32.5 mg ciprofloxacin DPI versus placebo (combined: n=156), and a power of >80% to detect the analogous difference between the 48.75 mg ciprofloxacin DPI and placebo groups (combined: n=120).
All efficacy variables were presented as summary statistics (arithmetic mean, SD, median, minimum and maximum for quantitative variables) and analysed by treatment group, dose of the study drug and corresponding placebo, and total, or combined by group. Frequency tables for qualitative data were provided. All statistical tests were two-sided and performed at the 0.05 significance level.
Change in FEV1 was analysed by three-way analysis of covariance, with the following factors: treatment group, macrolide use (no/yes), region (centres were pooled into three regions: northern USA and Canada, southern USA, and outside North America (Europe, Israel and Australia)). The dependent variable was FEV1 at EOT; baseline FEV1 served as a covariate. The following null hypotheses for treatment effect were tested in a three-step hierarchical testing scheme: ciprofloxacin DPI 32.5 mg=placebo 32.5 mg plus ciprofloxacin DPI 48.75 mg=placebo 48.75 mg (combined hypothesis); ciprofloxacin DPI 32.5 mg=placebo 32.5 mg and ciprofloxacin DPI 48.75 mg=placebo 48.75 mg (two single hypotheses); and ciprofloxacin DPI 32.5 mg=ciprofloxacin DPI 48.75 mg. Imputation for premature termination was by last observation carried forward. FVC and FEF25–75% end points were analysed in an analogous way.
Change in the density of P. aeruginosa in sputum was analysed by a log-linear model. The dependent variable was P. aeruginosa density (log10[colony-forming unit; CFU]+1]) in sputum at EOT (factors: treatment group, centre, macrolide use); pretreatment of P. aeruginosa density in sputum served as a covariate.
Time to antibacterial intervention for exacerbation was analysed by the Cox proportional hazards model, with baseline FEV1 as a covariate and the following factors: treatment, region and chronic macrolide use (no/yes).
Results
Subject disposition
A total of 288 patients were randomised. Of these, 286 received either study drug or placebo, and were included in the ITT and safety analyses sets. The first and last subject visits were 5 May 2008 and 25 January 2011, respectively. The main reason for discontinuation was AEs; these included pulmonary exacerbations requiring an antipseudomonal antibiotic (figure 1B). Baseline demographics were similar between groups (table 1). The overall mean age was 29.3 years with 16.8% of patients aged ≤20 years. Overall, 64% and 70% of patients in the ciprofloxacin DPI and placebo groups had received previous antipseudomonal maintenance therapy, respectively. Prior macrolide treatment status was similar for all groups (range 69.9–72.3%) except the 48.75 mg placebo group (54.3%). Owing to the 30-day exclusionary period for systemic/inhaled antibiotics, data on prior usage for specific agents were not systematically collected or available for all patients. Treatment compliance ≥80% occurred in 96.7% and 90.3% of participants in the ciprofloxacin DPI 32.5 and 48.75 mg groups, respectively, and in 90.8% and 85.7% of participants in the corresponding placebo groups. There were no obvious factors contributing to the slightly lower compliance rates in the placebo groups.
Baseline subject demographics and characteristics (intent-to-treat/safety population)
Primary objective
The primary end point testing did not demonstrate a treatment effect of ciprofloxacin DPI versus the corresponding placebo group or any difference between the doses in change in FEV1 from baseline to EOT in the ITT population (p=0.154; figure 2). Decreases from baseline were 0.01 L in both ciprofloxacin groups, versus 0.06 L and 0.07 L in the placebo 32.5 and 48.75 mg groups, respectively. However, the change in FEV1 from baseline to EOT for pooled ciprofloxacin DPI -treated patients was significantly less than for pooled placebo patients (p=0.02); the respective decreases were 0.02 L for the pooled ciprofloxacin DPI groups and 0.08 L ciprofloxacin DPI for the corresponding pooled placebo groups.
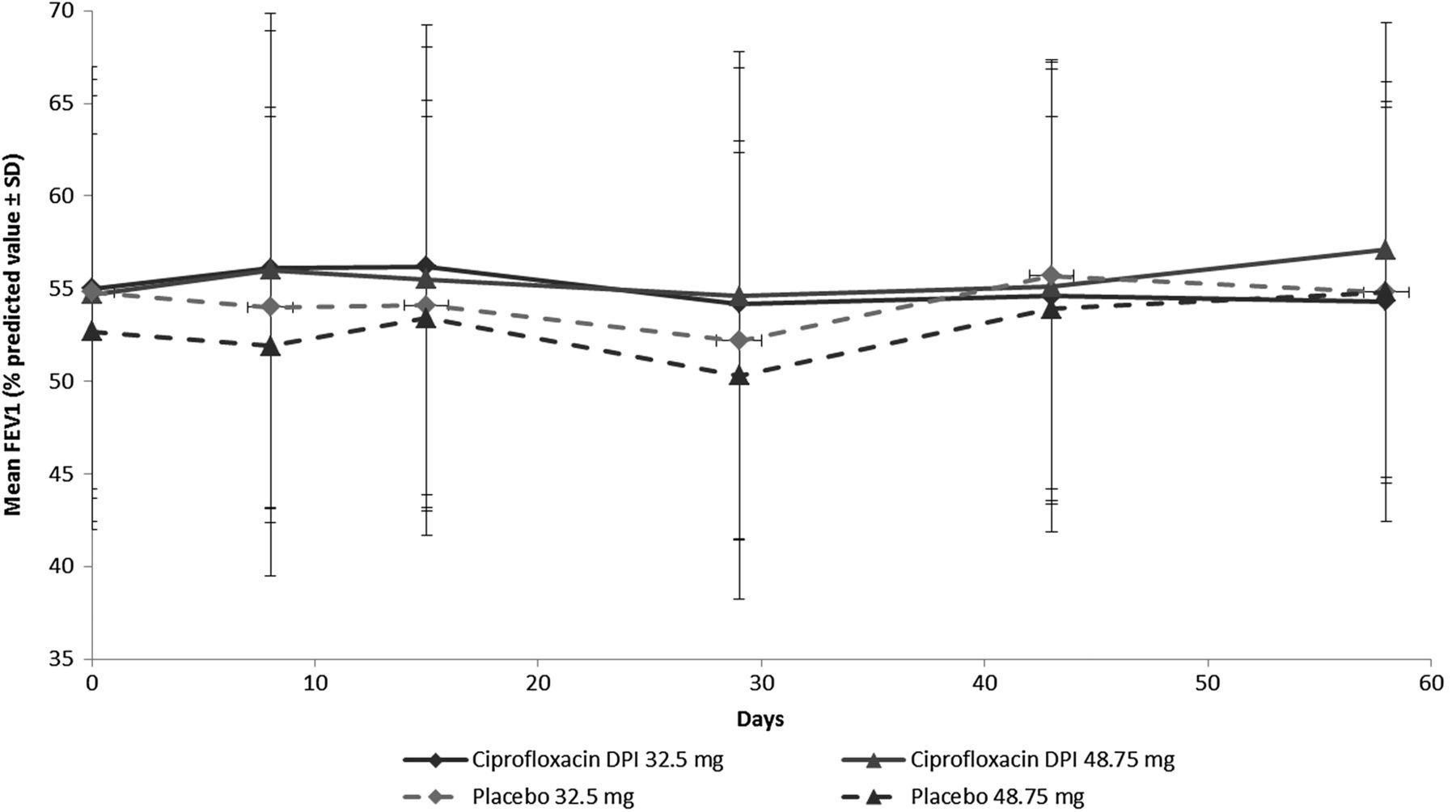
Time course of FEV1 (mean, % predicted) (intent-to-treat population) by study group. Empirical mean FEV1 is presented as percentage predicted value, with error bars representing SD. DPI, dry powder for inhalation; FEV1, forced expiratory volume in 1 s.
The time course of FEV1 values (empirical means) as a percentage of predicted values is shown in figure 2. In both ciprofloxacin DPI treatment groups, FEV1 increased from baseline to day 8, but declined after this time point to EOT. For the corresponding placebo groups, FEV1 decreased from baseline to EOT, except for a slight increase at day 15 in the 48.5 mg ciprofloxacin DPI placebo group. A slight increase in FEV1 was observed for all groups over the course of follow-up, with the exception of the 32.5 mg ciprofloxacin DPI group, in which FEV1 values remained relatively stable during this period.
Secondary objectives
Microbiology
Both doses of ciprofloxacin DPI resulted in a decrease in P. aeruginosa density during treatment, but this reduction was reversed after ending treatment (figure 3). P. aeruginosa density decreased from baseline values in both ciprofloxacin DPI treatment arms and the placebo 32.5 mg arm after treatment initiation. In the ciprofloxacin DPI 32.5 mg treatment arm, the mean bacterial density continued to decrease until day 15 (−1.0 log CFU/g), whereas a nadir was reached at day 8 for the 48.75 mg ciprofloxacin DPI treatment arm (−1.4 log CFU/g; figure 3). Although significant differences in P. aeruginosa density between ciprofloxacin DPI and placebo were observed at days 14–16 (p<0.001 and p=0.002 for the 32.5 and 48.75 mg doses, respectively), there were no significant differences in P. aeruginosa density at EOT (p=0.214 and p=0.388 for the 32.5 and 48.75 mg doses, respectively) and there were no significant changes in P. aeruginosa density in sputum from baseline at post-EOT follow-up visits. There was no difference between doses at any time point (p=0.726 at EOT). Log CFU reductions in mucoid P. aeruginosa were greater than the reductions in non-mucoid P. aeruginosa.

{kind=link}
{kind=link}
{kind=link}
Change in mean density of Pseudomonas aeruginosa in sputum (log10CFU/g) (intent-to-treat population) by study group. CFU, colony-forming unit; DPI, dry powder for inhalation.
There were no significant differences in the incidence of ciprofloxacin-resistant P. aeruginosa isolates in sputum at the 4-week follow-up visit. The MIC50/MIC90 values at baseline and EOT were 1/4 and 2/8 µg/mL for the ciprofloxacin DPI 32.5 mg group, and 2/8 and 2/16 µg/mL for the 48.75 mg group. Reductions in P. aeruginosa were similar in patients with baseline isolates that were susceptible (MIC ≤1 µg/mL), intermediate (MIC=2 µg/mL), or resistant (MIC ≥4 µg/mL) to ciprofloxacin (data not shown). The lack of correlation between reduction in P. aeruginosa and MIC susceptibility of organism to ciprofloxacin DPI is not surprising, considering the very high sputum concentrations of ciprofloxacin that can be achieved by inhalation of ciprofloxacin DPI.13
Respiratory end points
Exacerbations requiring any antipseudomonal intervention or hospitalisation occurred in 15/93 (16%) and 16/93 (17%) participants receiving ciprofloxacin DPI 32.5 and 48.75 mg, respectively, and in 13/65 (20%) and 10/35 (29%) participants receiving corresponding placebo. The study was not powered or designed to detect an effect of treatment on time to first exacerbation, and the differences observed between the ciprofloxacin DPI and placebo study arms were not statistically significant (32.5 mg vs placebo: p=0.464, HR 0.757, 95% CI 0.36 to 1.59; 48.75 mg vs placebo: p=0.143, HR 0.552, 95% CI 0.25 to 1.22).
The difference in mean percentage-predicted FEF25–75% from baseline to EOT in the ciprofloxacin DPI groups increased by 0.29% (pooled), 0.24% (32.5 mg) and 0.35% (48.75 mg). Conversely, the difference in mean percentage-predicted FEF25–75% in the matching placebo groups decreased by 2.0% (pooled), 2.1% (32.5 mg) and 1.8% (48.75 mg). There was no apparent treatment effect on FVC. No significant differences were seen in any of the other secondary end points (change in FEV1 from baseline to days 8 and 15, and at the 2-week and 4-week follow-up visits; change from baseline in FVC or FEF25–75% rate at all subsequent clinic visits; time to first pulmonary exacerbation requiring any antipseudomonal intervention or hospitalisation and microbiological end points discussed above).
Health-related quality of life
The 32.5 mg and pooled ciprofloxacin DPI groups both had significantly higher CFQR respiratory symptom subscale scores at EOT versus respective placebo groups (p=0.007 and p=0.019, respectively) in the ITT population. Differences were no longer significant by the 4-week follow-up. A weak but significant correlation was seen between CFQ-R respiratory symptom subscale score and FEV1 at EOT for the ITT (r=0.255) population (p<0.001). This correlation was still present in the ITT population at the 4-week follow-up (r=0.214, p=0.005).
Safety
There were no significant differences in type or incidence of TEAE by treatment group (all TEAEs p=0.115; table 2) in the safety population. A total of 43.5% of patients and 32% of patients receiving ciprofloxacin DPI and corresponding placebo, respectively, experienced a drug-related TEAE. Of the two ciprofloxacin DPI doses, the 32.5 mg dose appeared to be somewhat better tolerated; 37.6% of patients in this treatment group experienced a drug-related TEAE, compared with 49.5% of patients who received ciprofloxacin DPI 48.75 mg. The most common TEAEs in the ciprofloxacin DPI groups were dysgeusia and abnormal product taste, both of which were likely related to characteristics of the study medication (Ciprofloxacin is known for its bitter taste). Although the placebo formulation contained quinine to account for this, it is likely that the active drug and placebo formulations differed in bitterness or other taste attributes. Similar side effects related to taste were also reported in the ciprofloxacin DPI phase II trial in patients with non-CF bronchiectasis.17
Summary of treatment-emergent AEs (safety population)
Aside from three cases of haemoptysis that were considered to be related to the study drug, there were no new unexpected safety events reported during the study. Four participants had serious drug-related TEAEs. The incidence of treatment-emergent bronchospasm, defined as ≥15% drop in FEV1 following study drug administration, was low (0–3 participants per group) and there was no statistically significant difference by treatment group (p=0.741). No deaths occurred during the study.
Discussion
A significant need remains for the development of additional and easier-to-use inhalational antibiotics. Prolonged use of systemically administered antibiotics to patients with CF infected with P. aeruginosa is known to result in resistance to treatment and, therefore, diminished efficacy.23 To maintain efficacy on repeated administration, the strategy is usually to add another antibiotic to the treatment, which would have either an additive or a synergistic effect.24 Because the relevance of organism antibiotic sensitivity patterns has been questioned in recent years, the use of two antibiotics with different mechanisms of action to improve outcome is still common practice in most CF centres.25
Antibiotics administered through a DPI route have the potential for greater efficacy and safety, owing to targeted delivery of higher concentrations of the drug to the lungs and lower systemic exposure. Additionally, the more convenient method of administration (compared with traditional nebulisers) is likely to improve patient compliance.10 The primary end point of this study was to compare the change in FEV1 from baseline to EOT between patients receiving ciprofloxacin DPI and those receiving matching placebo. Based on the results of this study, the null hypotheses of no difference between the treatment groups could not be rejected.
Although the primary end point was not met, ciprofloxacin DPI was associated with some positive effects in analyses of the primary variable of lung function (FEV1), as well as on sputum bacterial load and health-related quality of life. In particular, there was a significant difference (p<0.05) between pooled ciprofloxacin DPI arms and pooled placebo arms in change in FEV1 from baseline to EOT. However, changes in FEV1 between 32.5 or 48.75 mg ciprofloxacin DPI and matching placebo, the prespecified primary study end point, did not achieve statistical significance. Both ciprofloxacin DPI arms transiently showed nominally significant results of reduced total bacterial load (P. aeruginosa) up to day 15 compared with matching placebo arms.
There was some evidence of both mucoid and non-mucoid P. aeruginosa reduction from baseline with ciprofloxacin DPI and, in the case of the former, these decreases were independent of resistance status at baseline. However, there was no sustained long-term decrease in the mean total bacterial load, as this change in P. aeruginosa density in the sputum from baseline was no longer statistically significant at EOT (ciprofloxacin DPI vs matching placebo; p=0.068). This finding has been noted in trials of other therapeutic agents, such as colistin.26
Significant improvements in health-related quality of life, assessed with the CFQ-R, were observed in the ciprofloxacin DPI 32.5 mg treatment group versus placebo as well as in pooled analyses at EOT (p=0.007 in the 32.5 mg ciprofloxacin DPI group vs matching placebo), although these improvements were not sustained at the 4-week follow-up visit (p=0.935 in the 32.5 mg ciprofloxacin DPI group vs matching placebo).
Several factors may have contributed to the primary end point not being met. Overall, the patients represented a heavily managed older population: approximately 77% of patients had received prior antipseudomonal maintenance therapy and mean age was 29.3 years; (16.8% of patients were aged ≤20 years), indicating a relatively advanced disease state in most patients.27 The limited progress in treatment over the last decade has reduced the potential to see clinically meaningful improvements in FEV1. Additionally, the lungs of older patients with CF are damaged to the point that significant improvements in FEV1 are less likely. Although suboptimal drug delivery or penetration could potentially have influenced the results of our study, data from phase I studies in CF participants indicate that twice daily ciprofloxacin DPI administration results in mean sputum ciprofloxacin concentrations of 34.9–149.7 mg/L, which are well above plasma levels achieved by systemic ciprofloxacin use (1.2–3.0 mg/L).28 ,29 In addition, scintigraphic data support high and reproducible lung deposition of approximately 50% in healthy study participants and in patients with non-CF bronchiectasis or chronic obstructive pulmonary disease.30 We therefore believe it is unlikely that poor drug delivery or penetration influenced the results. The overall reason for lack of FEV1 improvement in patients with CF treated with ciprofloxacin DPI is unknown, but this result demonstrates the complexity of CF inhalation studies and the possible need to consider alternative clinical end points in this population.31
Small airway occlusion by purulent mucus is common in the lungs of patients with CF, and this is likely to be more severe in patients with advanced disease. Disease severity may therefore influence distribution and location of the inhaled drug, as with any inhaled antibiotic. This, in turn, can affect the local concentration of ciprofloxacin.32 In addition to improved airway clearance before DPI administration, altering the dosing frequency may help to mitigate these challenges. In a study of levofloxacin inhalation solution, improved outcomes were observed with higher doses (240 mg twice daily vs 120 mg twice daily).33 In our study, however, there was no statistically significant difference between doses for the primary end point. The reasons for this are unclear, but may relate to a limited potential for improvement in this heavily pretreated patient population.
Overall, the safety parameters—type of TEAEs and their intensity, relationship to the study drug, seriousness and outcome—remained similar in all groups. There were three cases of haemoptysis considered to be related to the study drug, two of which were serious. However, it should be noted that haemoptysis is not an uncommon manifestation of pulmonary disease in CF. The 32.5 mg ciprofloxacin DPI seemed to be slightly better tolerated than the 48.75 mg ciprofloxacin DPI, with fewer cases of discontinuation due to AEs and fewer drug-related TEAEs. In both treatment groups, the incidence of cough was low. Inhalation of dry powder can result in cough, owing to the nature of the dosage form10; however, in this study, although the inhaled mass was greater than that inhaled with the TOBI Podhaler,12 a low incidence of cough was reported. This may be due to the fact that ciprofloxacin DPI is less osmotic than tobramycin; therefore, a lesser effect is being exerted on the lung epithelium.
Because of the short-term nature of this study, we are unable to speculate on the impact of ciprofloxacin DPI therapy on the effectiveness of future oral ciprofloxacin in this patient population. Studies with tobramycin solution for inhalation indicate that intermittent use in patients with CF results in modest increases in P. aeruginosa MIC values, but the increased MICs are not associated with clinically relevant effects.34 However, to date, no study has specifically examined the effect of inhaled antibiotics on subsequent systemic therapy with the same drug or drug class.
In conclusion, in patients with CF, twice daily treatment with ciprofloxacin DPI (32.5 or 48.75 mg) for 28 days did not result in significant improvements versus placebo in change in FEV1 from baseline to EOT, although some encouraging signs of efficacy were observed. It is possible that FEV1 is not the best end point for evaluating the efficacy of a short-term treatment on lung function in such a patient population. Ciprofloxacin DPI was safe and well tolerated; the 32.5 mg dose had a slightly more favourable overall tolerability profile than the 48.75 mg ciprofloxacin DPI dose. Additional studies assessing the efficacy of longer term (>28 days) treatment as indicated by alternative end points, as well as further investigations on optimal delivery of ciprofloxacin DPI in stable patients with CF, may be worth pursuing.
Acknowledgments
The authors would like to thank Barbara Hampel (formerly with Bayer HealthCare, Berlin, Germany) and Peter Reimnitz (formerly with Bayer Pharma, Wuppertal, Germany), for important intellectual contributions; and to acknowledge Fusion MD, Montreal, Canada, Highfield Communication, Oxford, England, and Kate Haynes of Chameleon Communications International, for providing medical writing services, with funding from Bayer Pharma AG. The authors would also like to thank the CF Foundation Therapeutic Network.
References
Footnotes
Collaborators The principal investigators in this study were: S Bell, H Greville, J Morton, P Robinson, D Serisier, P Thompson (Australia); AP Freitag, B Lyttle, K Palinder (Canada); T Pressler (Denmark); R Fischer, W Gleiber, D Staab, H Wirtz (Germany); L Bentur, H Blau, O Efrati, E Kerem, (Israel); A Hollsing, A Lindblad, L Mared (Sweden); M Carroll (UK); F Accurso, PJ Anderson, B Barnett, D Bisberg, S Boas, H Carveth, R Cohen, C Daines, Z Danov, SB Fiel, P Fornos, C Forseen, S Forsythe, D Froh, D Geller, R Gibson Jr, G Gong, D Hadjiliadis, D Homnick, M Howenstine, S Jain, ME Kleinhenz, N Kraynack, C Landon, D Layish, M Light, J Mcardle, B McWilliams Jr, K Meyer, S Miller, C Nakamura, S Nasr, MS Pian, A Prestridge, A Rao, S Reyes, J Rosen, J Royall, A Sannuti, D Schaeffer, G Sharma, A Stenbit, N Turcios, A Uluer, L Varlotta, R Vender, J Voynow, M Weatherly, J Wooldridge, W Yee, R Zanni (USA).
Contributors HLD and DS were involved in data acquisition. All the authors contributed to study design or were involved in data analysis and interpretation. All the authors participated in manuscript revision and approved the final version.
Funding Bayer HealthCare was the sponsor of this study, supplied the investigational agent, and provided funding for the clinical trial and for manuscript support by Fusion MD, Highfield Communication, and Chameleon Communications International.
Competing interests HLD has participated in studies funded by Bayer and has been a member of scientific advisory boards; EO, JA and MC are employees of Bayer.
Patient consent Obtained.
Ethics approval Ethics approval was obtained from Institutional Review Boards or Research Ethics Committees for all participating centres.
Provenance and peer review Not commissioned; externally peer reviewed.