Article Text

Abstract
Background Many patients with asthma have a T-helper type 2 (Th2) driven inflammation of the lung, whereas toll-like receptor 7 (TLR7) agonists, by inducing type I interferons, inhibit Th2 responses. In man, oral or parenteral TLR7 agonists can induce influenza-like symptoms through systemic induction of type I interferons. Design of a TLR7 agonist that is only active in the lung could reduce the risk of side effects and offer a new means for treating asthma. We developed a TLR7 agonist antedrug, AZD8848, to determine its local and systemic effects in preclinical models and man.
Methods In vitro cellular potencies for the TLR7 antedrug agonist, AZD8848, were determined along with pharmacokinetics and efficacy in a rat allergy model. Sputum and blood biomarkers were measured in single ascending and multiple ascending dose clinical studies following inhalation delivery of AZD8848 and tolerability assessed.
Results AZD8848 was potent in cellular assays and pharmacokinetics confirmed lack of systemic exposure to AZD8848. Weekly lung dosing in an animal model showed efficacy 26 days beyond the final dose. In healthy volunteers, AZD8848 was initially well tolerated with target engagement being demonstrated by induction of CXCL10 in sputum. A second inhaled dose, given 1 week later, amplified the systemic interferon signal in more than half the participants and resulted in significant influenza-like symptoms.
Conclusions The antedrug design restricted the direct actions of AZD8848 to the lung. However, the type I interferon induced locally by TLR7 spilled over systemically, limiting the utility of this inhaled antedrug approach.
Trial registration number NCT01560234, NCT01818869.
- Asthma
- Allergic lung disease
- Asthma Mechanisms
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
AZD8848 is a toll-like receptor 7 (TLR7) agonist antedrug, designed to have minimal systemic exposure, and has long-term efficacy in an animal allergy model.
Administration by inhalation in man shows tolerability following a single dose, but a second dose, 1 week later, results in the induction of flu-like symptoms.
The mechanisms behind this are investigated and further help to define the criteria necessary for this approach to become a future therapy for asthma.
Introduction
The prevalence of allergic diseases such as asthma is continuing to rise.1 ,2 Epidemiological studies have suggested a link between allergic disease and reduced microbial exposure of children leading to the ‘hygiene hypothesis’3–5 whereby reduced exposure to pathogens results in an immune deviation leading to an enhanced T-helper type 2 (Th2) response, characterised by the cytokines interleukin 4, IL-5, IL-13.6 Microbial effects are mediated through pathogen-associated molecular patterns that interact with the family of toll-like receptors (TLRs) expressed in innate immune cells, resulting in production of cytokines and expression of costimulatory molecules that modulate the adaptive immune response.7 Ten distinct TLRs have been identified8 and of these stimulation of either TLR7 or TLR9 in mouse disease models modulates many of the end points associated with allergy including suppression of goblet cell hyperplasia, reduction in Th2 cytokines, lung eosinophilia, lung inflammation and antigen-specific IgE.9–17
TLR7 agonists modulate Th2 responses in vitro through the action of interferon α (IFNα) on T cells as well as through the upregulation of the costimulatory ligand DLL-4,15 ,18 and via the downregulation of the Th2 transcription factor, GATA-3, by IFNα.19 In vivo, the importance of the role of type I IFNs, such as INFα, in TLR7-mediated immunomodulation of allergen-induced Th2 responses has also been established.16
TLR7 agonists have been administered in man for indications such as cancer and hepatitis C.20–22 While positive antiviral effects have been observed, these studies have all reported tolerability challenges, including influenza-like symptoms, which are associated with systemic cytokine induction. To attempt to reduce the systemic exposure of TLR7 agonism, AstraZeneca and Sumitomo Dainippon Pharma have collaborated to develop a series of 8-oxoadenine TLR7 antedrugs which can be administered topically to the lung and are designed to be rapidly metabolised to a less-active form on entry into the circulation.23 Although these compounds have short-term lung exposure, this is sufficient to preferentially activate the IFN pathway,18 leading to the local production of IFNα and modification of adaptive immune responses in animal allergy models.16
Further refinement of this chemical series culminated in AZD8848, which had characteristics suitable for progression into man. In this report, we document the in vitro and in vivo potency characteristics of AZD8848. Previous clinical studies with intranasal delivery of AZD8848 in patients with allergic rhinitis showed that it was safe and efficacious at tolerated doses.24 We now go on to describe the effects of the first ever reported inhaled delivery of a TLR7 agonist into the lung in man and report on the tolerability, pharmacokinetics and pharmacodynamics of AZD8848 administered by this route and reflect on the challenges facing this new treatment paradigm.
Methods
In vitro assays
Activity against human or rat TLRs was as described in Biffen et al's study.18 Peripheral blood mononuclear cell (PBMC) preparation and IL-5 inhibition assays are as described in Edwards et al's study.15 IFNα was determined following a 24 h incubation of PBMC with compound. In vitro plasma stability determinations were performed by methodologies described in Biffen et al's study.18
Studies in the Ovalbumin-sensitised Brown Norway rat
Animal studies were conducted in accordance with UK Home Office legislation under licence PPL40/2238; Procedure 5 and PPL40/2891 Procedure 2. Sensitisation, Ovalbumin (OVA) challenge and dosing of Brown Norway rats along with bronchoalveolar lavage (BAL) methodology is described in Matsui et al's study.16
For pharmacokinetic studies, following dosing, blood samples were collected into a solution of sodium fluoride (NaF), to inhibit esterase activity, and EDTA followed by extraction with water and methanol and analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). For lung sampling, tissue was excised and immersed in a solution of NaF and homogenised, extracted in methanol and analysed by mass spectrometry.
Clinical studies to evaluate inhaled AZD8848 dosing in man
Clinical studies were conducted in UK at the Quintiles Drug Research Unit, London, in accordance with the ethical principles in the Declaration of Helsinki and were approved by appropriate independent ethics committees. Written informed consent was obtained from all participants prior to the initiation of any study-related procedures.
Two randomised double-blind studies were conducted in healthy volunteers. Participants received a single dose of AZD8848 or placebo as a solution via a Spira nebuliser and the total dose was delivered over 10 inhalations in an ascending dosing design (NCT01560234). The doses were delivered to cohorts of six participants (4 active and 2 placebo) and each participant was dosed only once. Responses were assessed by a safety review committee before the next cohort was dosed. In the second study, participants were to receive four once-weekly inhaled doses of 30 µg AZD8848 (NCT01818869). The demographics of all participants are detailed in online supplementary table S1. Vital signs were monitored and blood samples taken for pharmacokinetic analysis, routine clinical safety markers, cytokine and RNA levels. Induced sputum samples were analysed for biomarkers. Adverse events (AEs) were noted by clinical staff and classified according to the Medical Dictionary for Regulatory Activities (MedDRA). The severity of the event was taken directly from the principal investigator's assessment.
Supplementary tables
Sampling for cytokine and gene expression measurements
Sputum was induced using a standard methodology.25 Cytokines were quantified in sputum and plasma on a Meso Scale Discovery multiplex platform according to the manufacturer's instructions. RNA from sputum cells and blood was isolated and analysed using standard methodology and reverse-transcriptase (RT) PCR was conducted using LifeTechnologies TaqMan gene expression assays performed on a Roche LightCycler 480.
Clinical pharmacokinetic measurements
Analysis of AZD8848 and its acid metabolite was performed using LC-MS/MS methods after solid phase extraction or extraction with ethanol.24
Results
AZD8848 is a potent, selective TLR7 agonist antedrug able to inhibit Th2 responses in vitro
We have developed AZD8848, which is an 8-oxoadenine acid methyl ester TLR7 agonist antedrug (figure 1A). In a bid to reduce systemic exposure, the compound was designed to be metabolised to its constituent acid in plasma, and has a half-life of <0.3 min in human and rat plasma (table 1). To further reduce systemic activation of TLR7, the acid metabolite, AZ12432045, is at least a 100-fold less potent than AZD8848 against recombinant human TLR7. AZD8848 has an EC50 of 4 nM in the induction of IFNα from human PBMCs and an IC50 of 0.2–1.0 nM for the inhibition of IL-5, irrespective of whether the T cells have been polyclonally stimulated with PHA or via antigen presentation (table 1). In both these assays, the acid metabolite was over 1000-fold less active. At concentrations up to 10 µM, AZD8848 has no activity against human TLR8 (table 1) or against any of the other human TLRs (data not shown), and shows good activity against rat TLR7, enabling its use in rat models.
Cellular potencies and plasma stability of AZD8848 and its acid metabolite, AZ12432045

Structure of AZD8848 and potency in recombinant human TLR human embryonic kidney (HEK) reporter cells. (A) The chemical structure of AZD8848 and (B) the activity of AZD8848 and its acid metabolite, AZ12432045, in HEK cells stably expressing human toll-like receptor 7 (TLR7). Activity was calculated from the release of secretory alkaline phosphatase, whose expression was linked to an NF-κB promoter, with the data normalised to the maximal induction produced by the reference TLR7 agonist, R848 (set to 100%). Incubation of cells with compound was for 24 h. Data show the mean±SEM from nine separate experiments (AZD8848) and from six separate experiments (AZ12432045).
Pharmacokinetics and efficacy of AZD8848 in the Brown Norway rat
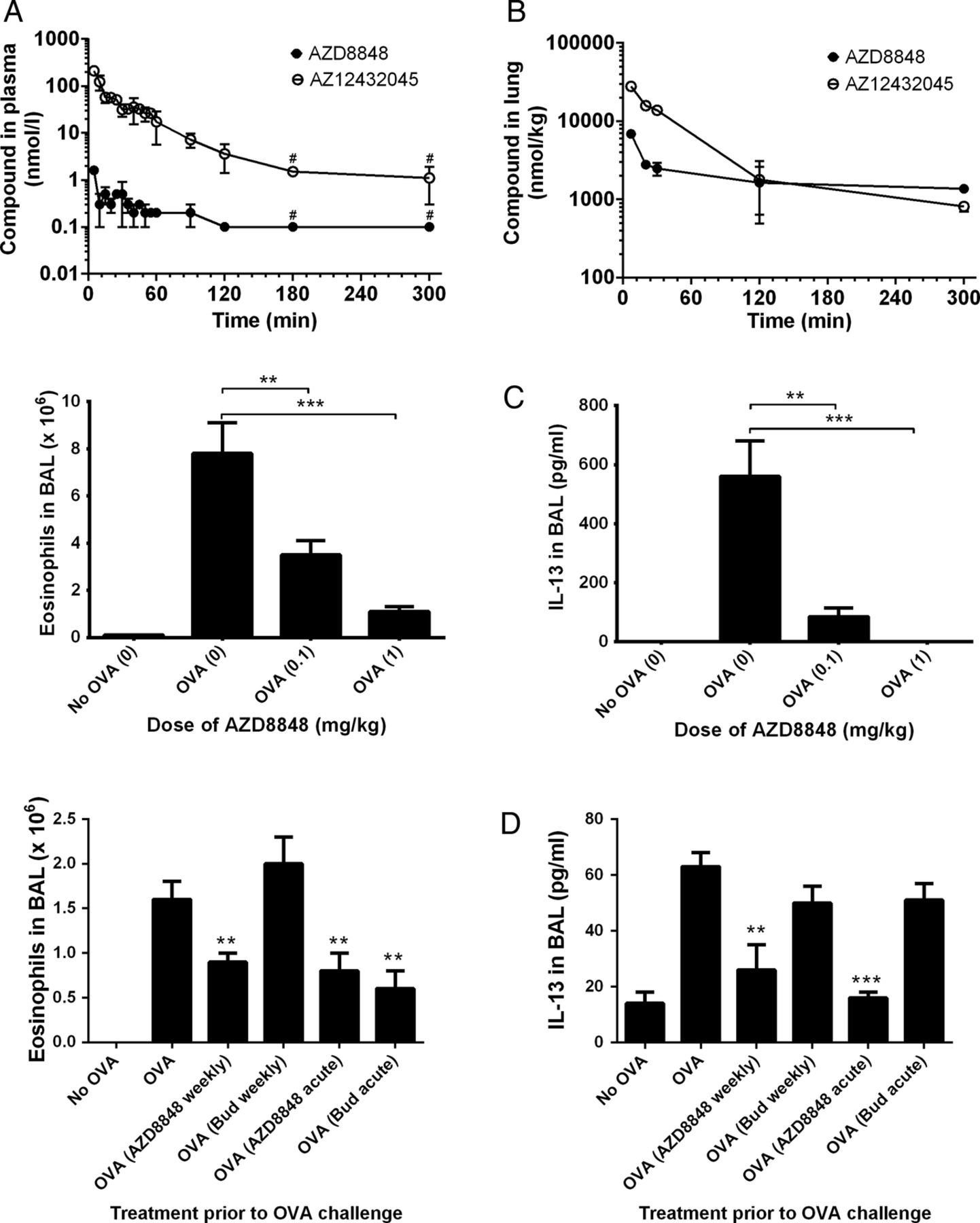
AZD8848 was dosed to the lung of Brown Norway rats and accurate determinations of the blood levels of AZD8848 were enabled by stabilising the samples with the esterase inhibitor, NaF. Under these conditions, the highest detected plasma concentration of AZD8848 was 2 nM at the 5 min time point (figure 2A), which reflects the very short in vitro half-life (0.2 min) in rat blood. The generation of the less-active acid metabolite in the blood was evidence of plasma esterase-mediated metabolism.

Pharmacokinetics and efficacy of AZD8848 in the Brown Norway rat. Brown Norway rats were dosed AZD8848 at 0.3 mg/kg intratracheally in a volume of 150 µL and blood samples taken at the indicated time points. Nine rats were dosed and at each time point samples from at least two animals were taken. Data for the plasma levels of compound (A) are presented as the mean of the determinations±SD or range and are representative of two such determinations. Following intratracheal administration, lung levels of AZD8848 and its acid metabolite were determined (B). At each time point, apart from the first, two animals were sacrificed and data are represented as the mean±range. The limits of detection for AZD8848 and AZ12432045 were 0.2 and 2 nmol/L, respectively. For the purposes of determining a representative value for each group of animals, any sample where the compound was undetectable was arbitrarily assigned a value of half that of the limit of detection. Data points including only such values are shown with a #. OVA-sensitised rats were dosed intratracheally with AZD8848, at the indicated doses (shown in brackets), 24 h prior to and 24 h after OVA challenge (C). Animals were sacrificed 48 h after the OVA challenge and the numbers of eosinophils or level of interleukin 13 in the BAL determined. The following rat numbers, in brackets, were used for each data point: sensitised only (5), intratracheally dosed (9). Data are representative of three studies. In further studies 14 days after OVA-sensitisation, Brown Norway rats were dosed topically with AZD8848 (1 mg/kg) or budesonide (1 mg/kg) via the intratracheal route on eight occasions at weekly intervals. Twenty-six days after the final dose of compound, animals were challenged with aerosolised OVA and 48 h later eosinophil number and IL-13 levels determined in the BAL (D). Ten animals were used for each data point. The effect of acute treatment with compound (24 h before and 24 h after OVA challenge) was also determined in groups of eight animals. The number of eosinophils or level of IL-13 in the BAL per animal was averaged across each treatment group and the result expressed as mean±SEM. All study groups were compared, with their respective OVA-stimulated control dosed with vehicle, using the Mann-Whitney U test; *p<0.05, **p<0.01, ***p<0.001.
In the lungs, the majority of the administered AZD8848 was cleared rapidly, with <10% of the dose remaining after 7 min (figure 2B). The lung level of AZD8848 declined slowly after this point and levels above 1000 nmol/kg were maintained for over 5 h. The differential in lung and plasma concentrations of AZD8848 demonstrates the successful antedrug design of AZD8848.
In the OVA-sensitised and challenged Brown Norway rat allergy model, OVA challenge resulted in a robust eosinophilia and induction of IL-13 in the BAL. These changes were suppressed in a dose-dependent manner following delivery of AZD8848 to the lungs 24 h before and 24 h after the OVA challenge (figure 2C).
A single dose of AZD8848 resulted in a duration of effect lasting for up to 5 days (results not shown). Following eight once-weekly doses of AZD8848 in the Brown Norway rat model, a subsequent OVA challenge 26 days after the last dose of AZD8848 resulted in significant inhibition of BAL eosinophilia and IL-13 production, which was as efficacious as acute dosing with AZD8848, 24 h before and 24 h after OVA challenge (figure 2D). Whereas acute dosing with budesonide demonstrated efficacy, a protocol of once weekly dosing with budesonide did not result in any prolonged duration of effect, unlike that observed with AZD8848.
Pharmacokinetics following inhalation dosing of AZD8848
We performed a single ascending dose study in healthy volunteers to assess target engagement and tolerability. Following inhaled administration, AZD8848 was only directly measurable in a small number of plasma samples, all within 10 min of dosing (data not shown). The maximal level of AZD8848 detected in plasma was <0.1 nmol/L, which is 1000-fold below the in vitro EC50 for stimulation of human TLR7. The clearance of AZD8848 from the lung was therefore monitored indirectly through levels of its immediate hydrolysis product, AZ12432045, which peaked within 10 min of inhalation of AZD8848 (figure 3A). The mean maximum plasma concentration of AZ12432045 increased with dose, reaching 1.7 nmol/L at the highest dose, which was at least 1000-fold below its activity in human cellular assays. Thus, the antedrug design of AZD8848 was successful in preventing the systemic circulation being exposed to biologically significant levels of the TLR7 agonist.
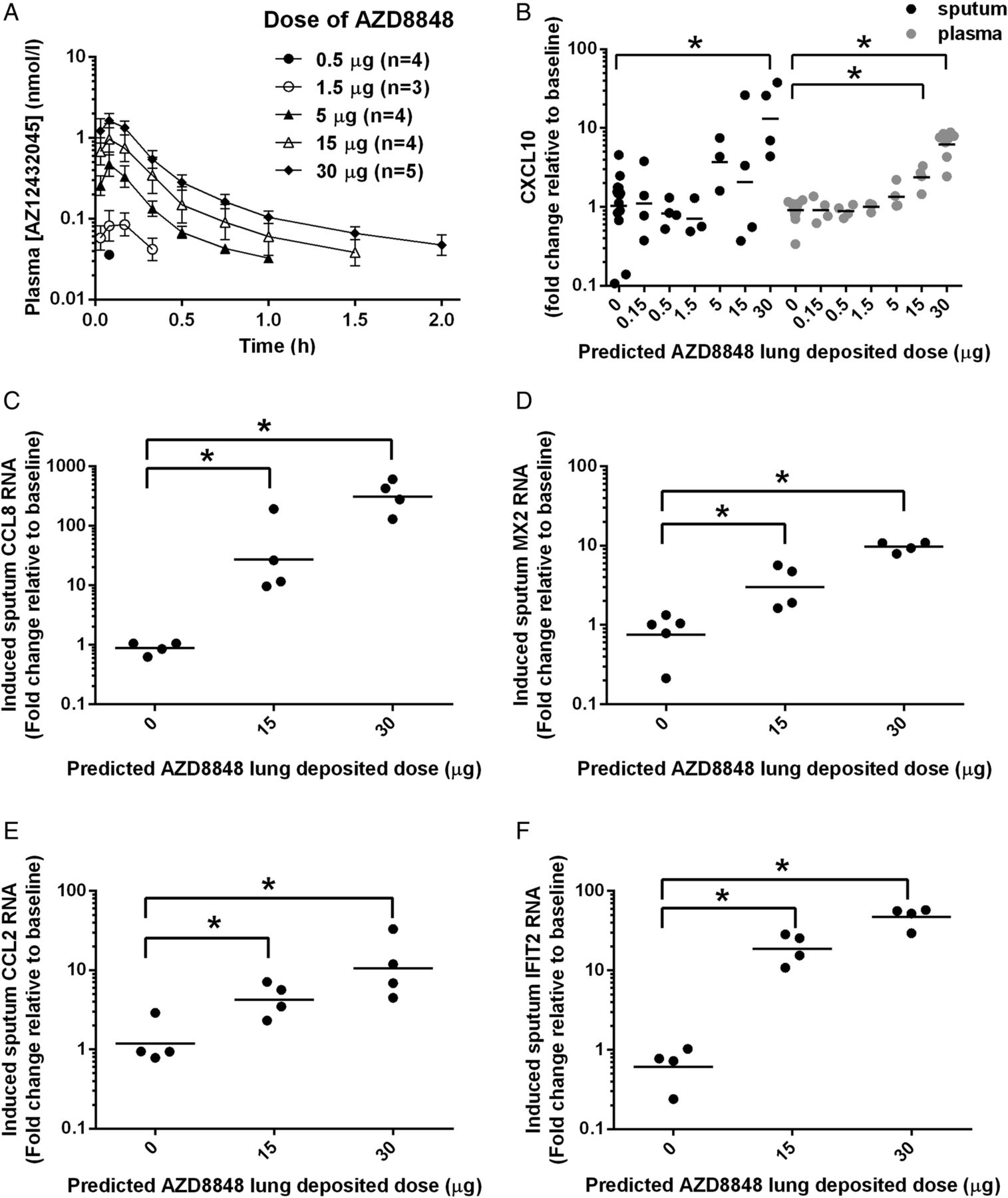
Pharmacokinetics and biomarker induction following a single inhaled dose of AZD8848 in man.(A) Plasma concentration-time profiles of AZ12432045, the acid metabolite of AZD8848, following a single predicted lung-deposited dose of AZD8848 as indicated in the key. The predicted lung-deposited dose was assumed to be 63% of the dose delivered from the nebuliser. During sample preparation, any trace amounts of AZD8848 were hydrolysed by esterases in the plasma. All cohorts were administered by nebulisation taken in 10 breaths as described in Methods section. No data are shown for cohort 1 (0.15 µg) as levels of AZ12432045 were below the level of detection. Values shown are geometric means (±SD) from 3 to 5 individuals in each cohort. (B) CXCL10 was determined in induced sputum (black) and in plasma (grey) of healthy volunteers for the indicated predicted lung-deposited dose of AZD8848. The fold change at 24 h postdose from baseline for individual participants is shown. Geometric mean is shown in each cohort and significance relative to placebo (p<0.05) is indicated on the basis of analysis of covariance (ANCOVA) analyses with baseline as a covariate. RNA changes for CCL8 (C), MX2 (D) CCL2 (E) and IFIT2 (F) were determined in induced sputum cells using quantitative RT-PCR. Geometric means are shown in each cohort and significance relative to placebo, *p<0.05, is indicated on the basis of two-sample Student t-tests.
Single inhaled doses of AZD8848 are tolerated and induce local and systemic IFN responses
The IFN-inducible chemokine, CXCL10, is a sensitive measure of TLR7 activation20 ,26 and was monitored in the sputum and in the blood following inhalation of AZD8848. Increased production of CXCL10 in the lung, as measured in the induced sputum collected 24 h after dosing, was statistically significant at the 30 µg dose level (figure 3B). The RNA levels of other IFN regulated genes, CCL8, MX2, CCL2 and IFIT2, showed significant dose-dependent increases in sputum cells following 15 µg and 30 µg doses of AZD8848 (figure 3C–F). However, despite the lack of systemic AZD8848, increases in levels of plasma CXCL10 protein were detected, which were statistically significant at the 15 µg and 30 µg doses (figure 3B). All dose levels of AZD8848 were well tolerated and the reported AEs were mostly mild (see online supplementary table S2).
Second inhaled doses of AZD8848 are less well tolerated and correlate with an exaggerated plasma CXCL10 response
The 30 µg dose of AZD8848 gave significant induction of CXCL10 protein and was chosen to be dosed to healthy volunteers once weekly for 4 weeks. However, after the second dose, four of the six individuals reported influenza-like symptoms. Symptoms started 2.5–4.5 h after dosing and, in two cases, temperatures of 38.3°C and 39.4°C were recorded 6–8 h postdose (table 2). Participants experiencing influenza-like symptoms were treated with paracetamol and symptoms had resolved by 24 h postdose. As a consequence of the number of AEs following the second dose, the trial was stopped. Follow-up blood samples were taken to explore the mechanism behind this abrupt change in tolerability.
The number of participants reporting AEs in the MAD study
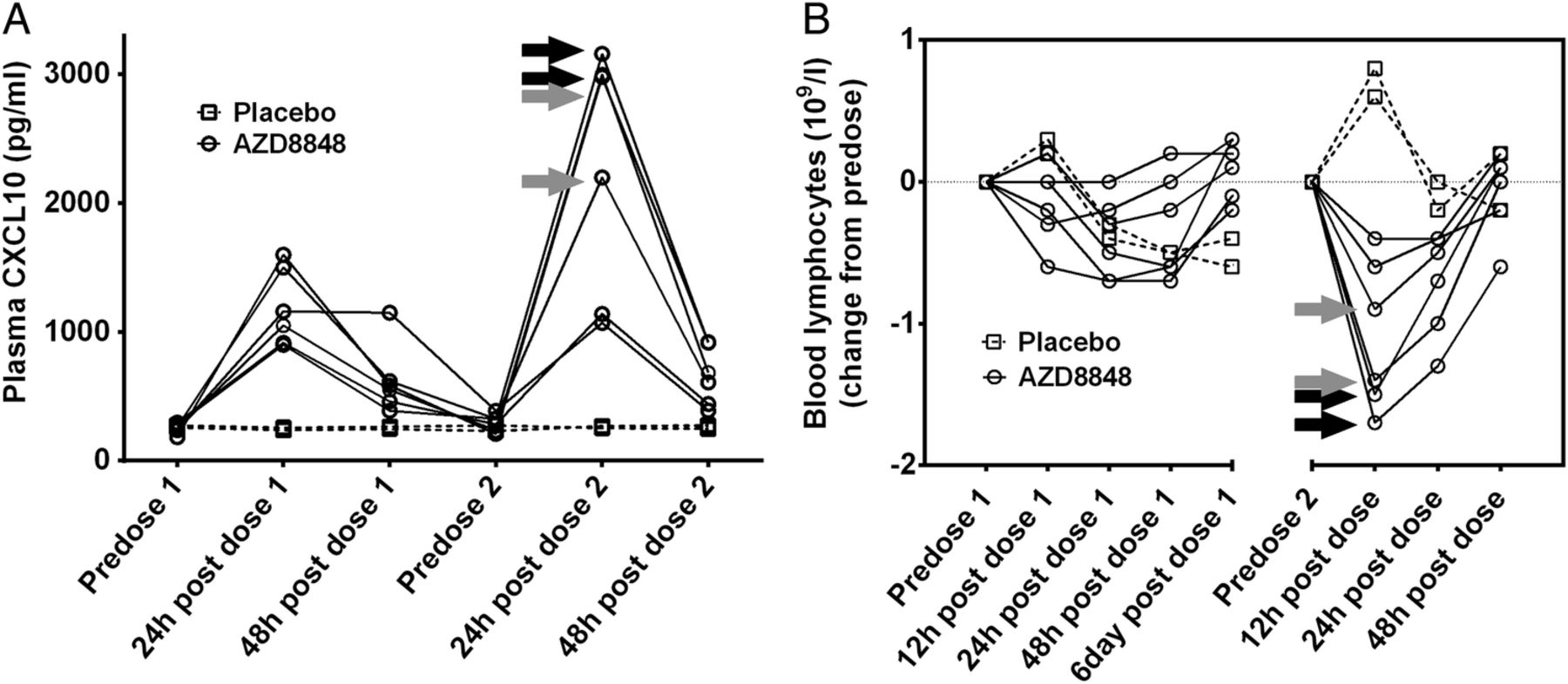
AZD8848 (30 µg) induced a significant elevation in the level of plasma CXCL10 24 h after dosing compared with placebo (analysis of covariance (ANCOVA) values of p=0.0001 after the first dose and p=0.0037 after the second dose). The induction after the second weekly dose was statistically greater than the levels achieved after the first (within-subject paired Student t test, p<0.05). Data for each individual participant are shown in figure 4 and the plasma CXCL10 response in four of the six individuals was greater following the second dose of AZD8848 compared with the first (figure 4A), even though it had returned to near baseline values in the intervening period. The participants with the greater increases in the level of CXCL10, 24 h after the second dose, corresponded to the participants experiencing influenza-like symptoms. The two participants who did not report influenza-like symptoms gave a CXCL10 response similar to that seen after the first dose.

Plasma CXCL10 and blood lymphocyte counts following a second inhaled dose of AZD8848. CXCL10 in plasma (A) and lymphocytes in blood (B) were determined in healthy volunteers receiving two 30 μg doses of AZD8848 1 week apart. Data from individual participants are shown as separate lines, treatment as solid lines, and placebo as broken lines. Arrows indicate individual participants who experienced severe (black) or moderate (grey) influenza-like symptoms in the preceding period. Between-treatment and intrasubject comparisons for CXCL10 levels are described in the text.
There was a modest but transient drop in blood lymphocytes after the first dose of 30 µg AZD8848 (figure 4B). This effect was more pronounced and occurred more rapidly after the second dose. Those individuals showing the greatest reduction in circulating lymphocyte numbers were also the ones experiencing flu-like symptoms.
Lack of correlation of other biomarkers with AEs
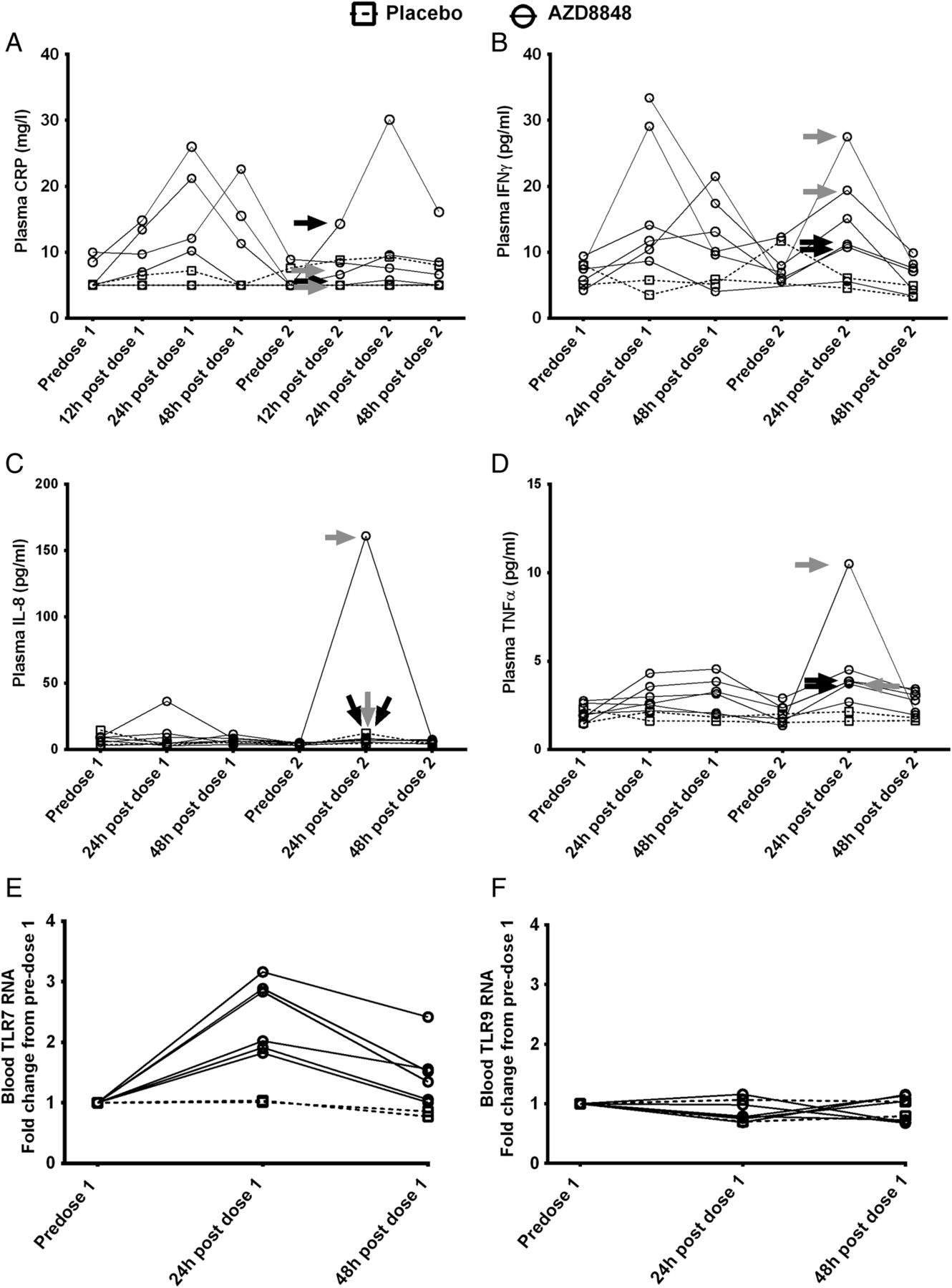
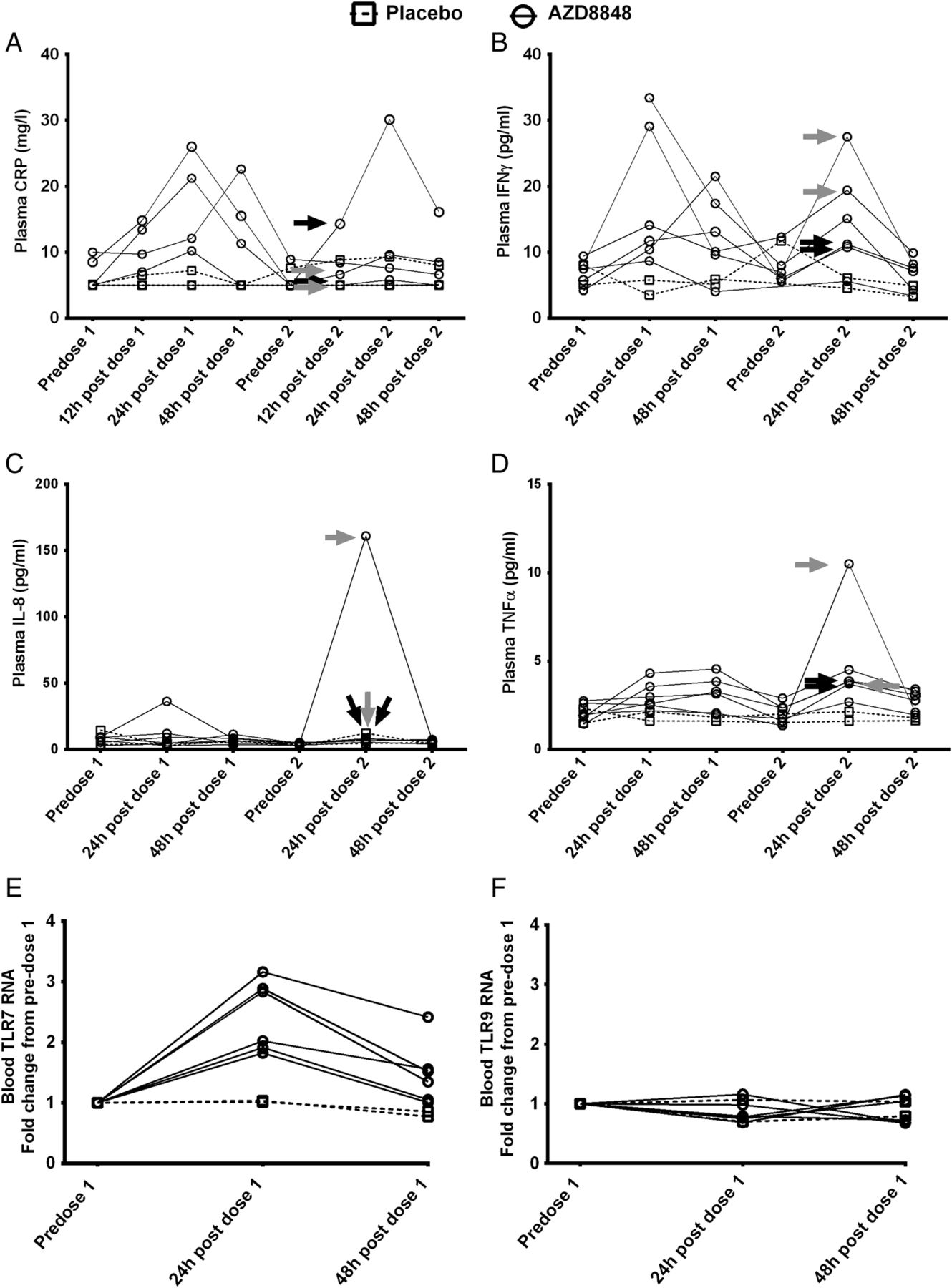
After the first and second weekly dose of AZD8848, tumour necrosis factor α (TNFα) and IFNγ levels in the plasma showed similar transient increases in levels 24 h postdosing, which were returning towards baseline at 48 h postdosing (figure 5B, D). C reactive protein (CRP) and IL-8 (figure 5A, C) showed increases in some individuals but not others. In contrast to CXCL10, none of these cytokines showed comparably higher levels after the second dose, and nor did they correlate with influenza-like symptoms. It has been reported for the stable TLR7 agonist PF-487869120 that enhanced responses on repeated dosing may result from increased expression of TLR7. Expression of TLR7 RNA in blood showed a transient increase 24 h after the first dose of AZD8848, although this had returned to near baseline 48 h after the first dose (figure 5E, F). In contrast to TLR7, TLR9 RNA was not increased.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Monitoring the induction of pro-inflammatory mediators and toll-like receptors (TLRs) following inhalation of AZD8848. The mediators (A) C reactive protein (CRP), (B) interferon γ (IFNγ), (C) interleukin 8 (IL-8) and (D) tumour necrosis factor α (TNFα) were measured in plasma as described in Methods section in healthy volunteers receiving two 30 μg doses of AZD8848 1 week apart. Data from individual participants are shown as separate lines, treatment as solid lines, and placebo as broken lines. Arrows indicate individual participants who experienced severe (black) or moderate (grey) influenza-like symptoms in the preceding period. Changes in (E) TLR7 and (F) TLR9 RNA levels in blood were determined in healthy volunteers receiving a 30 μg dose of AZD8848 and samples were taken at the time points indicated. Data from individual participants are shown, those receiving placebo as a broken line, those receiving AZD8848 as a solid line. In view of the small numbers of participants, no statistical testing was performed between treatment groups.
Discussion
TLR7 agonists suppress Th2 responses9–16 and as atopic asthma is associated with an enhanced Th2 biological drive, TLR7 agonists could be developed as a new treatment paradigm for this disease. However, the generation of ‘influenza-like’ symptoms resulting from either systemic TLR7 activation or elevated systemic levels of TLR7-induced mediators has been reported in TLR7 clinical programmes.20–22 We sought to restrict the effect of TLR7 agonism to the lung by generating a series of TLR7 agonist antedrugs,18 ,23 which culminated in AZD8848, a TLR7-selective agonist with good potency and a human plasma half-life of <0.3 min. In addition, the acid metabolite to which it is directly converted is at least 100-fold less active than the parent compound. These features, coupled with the pharmacokinetic data in man, demonstrated that the antedrug design of AZD8848 was successful in limiting the functional activity of AZD8848 to the lung where it was dosed.
AZD8848 was efficacious at reducing OVA-induced inflammation in the lung in the Brown Norway rat model, and following eight weekly doses to the lung AZD8848 had an effect lasting at least 26 days after the last dose. Thus, despite the short duration of exposure following each dose, which resulted from the antedrug mechanism, repeated pulses of AZD8848 stimulation were able to induce a long-standing state of inhibition of allergic inflammation. An extended duration of action has also been observed following repeated dosing of the TLR9 agonist, 1018 ISS, in a murine model of allergic airway inflammation, where 12 weekly doses induced a suppression of allergic Th2 parameters which was maintained for a further 15 weeks.17 Such a prolonged effect suggests some modulation of the adaptive immune response and in the case of 1018 was associated with induction of Treg responses.
Inhalation of AZD8848 to the lungs in healthy volunteers demonstrated that a single dose of up to 30 µg was well tolerated and was associated with an increase in IFN-stimulated genes and protein in sputum. The antedrug nature of AZD8848 led to its rapid metabolism in plasma and prevented it achieving systemic levels required for biological activity. Yet, despite this, CXCL10 was elevated in plasma after inhalation of AZD8848. Our data do not support circulating levels of AZD8848 inducing CXCL10 systemically, and most probably the CXCL10 originated from TLR7 activation within the lung. Stimulation of plasmacytoid dendritic cells (pDCs) within the lung would induce type I IFN and this would, in turn, induce CXCL10, which could spill over into the systemic circulation. Alternatively, locally produced type I IFN itself could enter the circulation and induce CXCL10 systemically. However, although the antedrug design prevented AZD8848 from having direct systemic activity, the mediators it generated within the local tissue were not similarly confined.
Despite the good tolerability of a single inhaled dose of AZD8848, a second dose of 30 µg, given 1 week later, resulted in four of the six participants reporting influenza-like symptoms. In these individuals, there was also an amplified response in plasma CXCL10 and a more profound decrease in circulating lymphocytes. Type I IFNs are known to cause influenza-like symptoms,27 mediate a decrease in blood lymphocytes28 and stimulate genes such as CXCL10.20 ,26 Such effects are consistent with the scenario that the effects observed after the second dose are due to an exaggerated type I IFN response in the lung, leading to an increased systemic spillover in four of the participants. Previously, when AZD8848 was administered to the nasal cavity, no enhancement in the IFN-inducible gene, IL-1 receptor antagonist, in plasma was observed over the five weekly doses.24 Thus, in contrast to nasal dosing, the first dose given directly to the lung somehow primes for a greater effect with the second dose. However, this did not occur in all participants, and currently we have no means of identifying which participants will respond in this way. Following inhalation of AZD8848, of the other mediators measured, only plasma IFNγ and TNFα were consistently quantifiable and in both cases their levels of induction showed no amplification between the first and second doses. This indicated that their levels were not linked to the deterioration in tolerability.
Independent modulation of mediators following TLR7 activation is consistent with in vitro experiments showing that short-term exposure to a TLR7 agonist preferentially activated the type I IFN pathway relative to that of the NF-κB pathway.18 Since plasmacytoid dendritic cells are the primary source of type I IFN following TLR7 stimulation, this may indicate that the priming event caused by the first dose of AZD8848 in these individuals impacts on the pDC axis. The cellular and molecular mechanisms by which the IFN pathway is primed and amplified remain unclear. Possibilities include enhanced expression of TLR7 on the pDCs within the lung, an increase in the number of pDCs within the lung or a combination of both. A small increase in the expression of circulating TLR7 mRNA following administration of a TLR7 agonist has been observed in this study and others.20
In contrast, in a murine study, intravenous administration of a TLR7 agonist resulted in the down modulation of TLR7, which corresponded to a decreased induction of IFNα following a subsequent dose of a TLR7 agonist and reduced efficacy when dosing was twice weekly compared with weekly.29 In this study, the tolerising effect on TLR7 was independent of type I IFN and may well reflect differences in the dose and dosing regimen. In our study, weekly dosing was with a TLR7 agonist at doses that were only just showing significant induction of IFN-inducible markers, whereas the murine study was using a dose that was giving near maximal TLR7 stimulation. It is possible that the prolonged receptor stimulation or additional mediators induced in the murine study contributed to the down modulation of TLR7. In our study, the elevation in TLR7 mRNA had returned to baseline when the second dose was administered, although we have no data on the actual expression of TLR7 protein over this period. Additionally, the changes in TLR7 RNA levels observed after the first dose did not predict those individuals giving the amplified CXCL10 response after the second dose. Influx of pDCs into the lung following TLR7 antedrug administration to the lung have been observed.16 However, to resolve whether pDC numbers or TLR7 expression remain elevated 1 week after an initial dose of AZD8848 in the lungs of man would require further sampling of the sputum or even lung biopsies.
Previous studies identified no tolerability issues with multiple weekly dosing of AZD8848 to the nose.24 This tolerance may relate to the area of tissue exposed to AZD8848. Inhalation to the lung would expose substantially more tissue than dosing to the nose and, correspondingly, more pDCs could be activated. Given the well-perfused state of the lung, any enhanced production of type I IFN resulting from the pDC activation could readily transfer to the circulation where it could lead to influenza-like symptoms.
In preclinical models, multiple dosing with a TLR7 agonist results in the long-term modification of allergic responses far beyond the period of dosing (this study and Xirakia et al's study11). The possible translation of this sustained effect to human asthmatic disease opens up the exciting possibility of long-term disease modification, perhaps even remission, without the need for continuous medication. However, although the first dose was well tolerated, we found that a second lung dose in healthy volunteers resulted in an unacceptable increase in side effects in some participants. Since this was seen at a dose of AZD8848 that was at the threshold of target engagement in the target tissue, the likelihood of establishing a dose with weekly dosing that is both efficacious and tolerable for patients with mild asthma appears low for this class of TLR agonist antedrug. Whether less-frequent dosing would resolve this issue would depend on further preclinical data demonstrating efficacy with such a dosing regimen. Nevertheless, the prospect of inducing a long-term disease modification in asthma through this type of mechanism remains enticing, provided the consequences of TLR activation can be suitably confined to the lung.
Acknowledgments
The authors are grateful to Dr Haruo Takaku (Sumitomo Dainippon Pharma) and Dr Ash Bahl for their scientific leadership resulting in the selection of AZD8848 and to Dr Sam Lindgren and Dr Paul Rugman for their input into the design of the clinical studies. They wish to thank Dr Andrew Leishman for data on IL-5 inhibition in the human PBMC Der p 1 assay; Dr Amanda Berry for support in delivering the preclinical pharmacokinetics and Jim Britt for assisting in the in vivo studies. They are grateful to Dr Marita Olsson and Dr Hong-Lin Su for performing statistical analyses. They are also indebted to the principal investigators, Professors James Ritter and Tim Mant, for the SAD and MAD studies, respectively.
References
Footnotes
Contributors SD was responsible for plans for biomarker studies and interpretation of results; intellectual input into the design of clinical trials and interpretation of data from the trials. MB was responsible for establishing in vitro assays, testing compounds and interpreting data; intellectual input into design of clinical trials and interpretation of data from the trials. JM was responsible for plans for biomarker studies, running the assays and interpretation of results. JB was responsible for designing and running in vivo assays. SA was responsible for design of studies to monitor pharmacokinetics in man and analysis and interpretation of data. AA was responsible for design and running of clinical trials and interpretation of data from the trials. MK contributed to the design of the clinical studies and interpretation of the results. DK was involved in the interpretation of results and was responsible for drafting of the manuscript.
Funding These studies were funded by AstraZeneca.
Competing interests All authors are either current or former employees of AstraZeneca.
Ethics approval National Research Ethics Service, London UK.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.