Article Text

Abstract
Introduction Idiopathic pulmonary fibrosis (IPF) is a chronic and fatal disease of unknown cause characterised by progressive fibrotic formation in lung tissue. We hypothesise that disrupted metabolic pathways in IPF contribute to disease pathogenesis.
Methods Metabolomics of human IPF was performed using mass spectroscopy (IPF lung=8; donor lung=8). Gene expression of key metabolic enzymes was measured using microarrays. Of the 108 metabolites whose levels were found altered, 48 were significantly increased, whereas 60 were significantly decreased in IPF samples compared with normal controls.
Results Specific metabolic pathways mediating the IPF remodelling were found with a downregulated sphingolipid metabolic pathway but an upregulated arginine pathway in IPF. In addition, disrupted glycolysis, mitochondrial beta-oxidation and tricarboxylic acid cycle, altered bile acid, haem and glutamate/aspartate metabolism were found in IPF samples compared with control.
Conclusions Our results show alterations in metabolic pathways for energy consumption during lung structural remodelling, which may contribute to IPF pathogenesis. We believe that this is the first report of simultaneously and systemically measuring changes of metabolites involving nine metabolic pathways in human severe IPF lungs. The measurement of the metabolites may serve in the future diagnosis and prognosis of IPF.
- interstitial fibrosis
- lung transplantation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
To study the metabolic changes in idiopathic pulmonary fibrosis, which may contribute to disease pathogenesis.
The results show specific metabolic pathway and genetic profile alterations in the advanced stage of the human idiopathic pulmonary fibrosis lung.
The results of this study support a greater understanding of the metabolic basis of pathogenesis of idiopathic pulmonary fibrosis formation, and the metabolite changes in idiopathic pulmonary fibrosis lungs may represent a group marker for the prediction of the development of idiopathic pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive disease in which healthy lung tissue is replaced by scar tissue, resulting in impaired air exchange in the lungs. The incidence of IPF has recently been estimated to be between 14 and 42.7 per 100 000 and with median survival time estimated to be 2.5 to 3.5 years.1 Lung transplantation remains the only therapy that prolongs survival in advanced IPF.2
The exact cause of IPF is unclear, but it is thought that alveolar epithelial damage, and subsequent dysregulated wound repair, may lead to progressive lung fibrosis.2 Factors that damage the epithelium, such as smoking, wood dust and epithelial endoplasmic reticulum stress, have been linked to IPF.1 3 In addition, multiple wound repair signalling pathways, such as the coagulation cascade, have been found to be dysregulated in IPF.4 Stress to the epithelium also releases transforming growth factor-beta (TGF-beta), which can act on fibroblasts to increase production of profibrotic factors such as collagen, fibronectin and alpha-smooth muscle actin. TGF-beta has also been shown to induce apoptosis in epithelial cells both in vitro and in the bleomycin-induced fibrosis model.5 Recent studies in IPF have revealed exciting results in cellular pathways related to epithelial injury, oxidative stress, immunity and energy metabolism, which provide insight into IPF pathogenesis. Proteomic analyses6 in human IPF lung samples found upregulated levels of proteins relevant to endoplasmic reticulum stress and oxidative stress, with downregulated levels of antiapoptotic factors. An analysis of the peripheral blood transcriptome in patients with IPF revealed changes in pathways related to host immunity.7 Finally, a lung inflammation and fibrosis model with mice exposed to silica found altered cell energy metabolism and amino acid metabolism leading to collagen synthesis through proline and hydroxyproline.8
Metabolomics is an exciting tool to study cellular mechanisms that underlie disease progression, but there have been few systematic measurements of metabolic products in IPF. Our group’s published9 data in patients with pulmonary arterial hypertension (PAH) showed that abnormal levels of metabolites involved in glycolysis and fatty acid oxidation ultimately affected bile acid production in the lung. In the current study, we studied the metabolic changes in patients who developed severe IPF. Our results show alterations in metabolic pathways for energy consumption during lung structural remodelling, specifically, involving nine metabolic pathways in human severe IPF lungs. Measurement of these metabolites may serve as a future strategy to help in the diagnosis and prognosis of IPF.
Materials and methods
Patients and tissue collection
The biochemical profiles were determined in human lung tissue and compared across eight normal (47±15 years of age, four females) and eight patients with IPF (59±8 years of age, two females). All patients provided informed consent for the study, which was approved by the institutional Human Tissue Committee and Research Ethics Board of the University Health Network, University of Toronto. Eligibility criteria included patients with end-stage IPF who went through lung transplantation. Lung samples were collected from the recipient lung at the time of lung transplantation. Control lung samples were collected from normal tissue of patients with cancer undergoing surgical lobectomy. The samples of lung tissues were frozen and stored in −80°C prior to sample analysis. Pulmonary arterial pressure was measured by right heart catheterisation performed for clinical care with mean pulmonary artery pressures of 32±12 mm Hg, pulmonary capillary wedge pressure of 7±3 mm Hg and 6 min walking distance of 295±94 m. The lung function analysis of patients with IPF showed forced expiratory vital capacity of 54%±18% predicted (pred), diffusing capacity of the lung for carbon monoxide 41%±15% pred and total lung capacity 61%±14% pred. All patients provided written informed consent, in accordance with the Declaration of Helsinki, for research protocols approved by the institutional review boards of the University Health Network.
The details of metabolomics and microarray analysis are given in the online supplementary data.
Results
IPF lung samples displayed significant changes in metabolites involved in the metabolism of sphingolipid, arginine, energy (including glucose, fatty acid and citric acid metabolism), bile acid, haem and glutamate/aspartate metabolic pathways. We also found significant changes in expression levels of genes encoding enzymes that control and regulate affected metabolic pathways.
Reduced sphingolipid metabolism
Sphingolipids are components of plasma membranes and have been linked to cellular proliferative pathways, which may implicate them in lung structural remodelling in IPF. We found decreased levels of the metabolites sphinganine, sphingosine and sphingomyelin in patients with IPF compared with control (figure 1). The microarray data also showed downregulated delta(4)-desaturase, sphingolipid 1 (DEGS1) and sphingomyelinases (SMPD1 and SMPD4), which point to reduced ceramide production. We also found downregulated alkaline ceramidase 3 (ACER3) and sphingosine kinase 1 (SPHK1), suggesting reduced production of sphingosine and sphingosine 1-phosphate (S1P), respectively. Finally, downregulated expression of S1P receptors, sphingosine 1-phosphate receptor 1 (S1PR1) and S1PR4, indicates reduced S1P signalling. Taken together, our data suggest decreased metabolism of sphingolipids in IPF lungs.
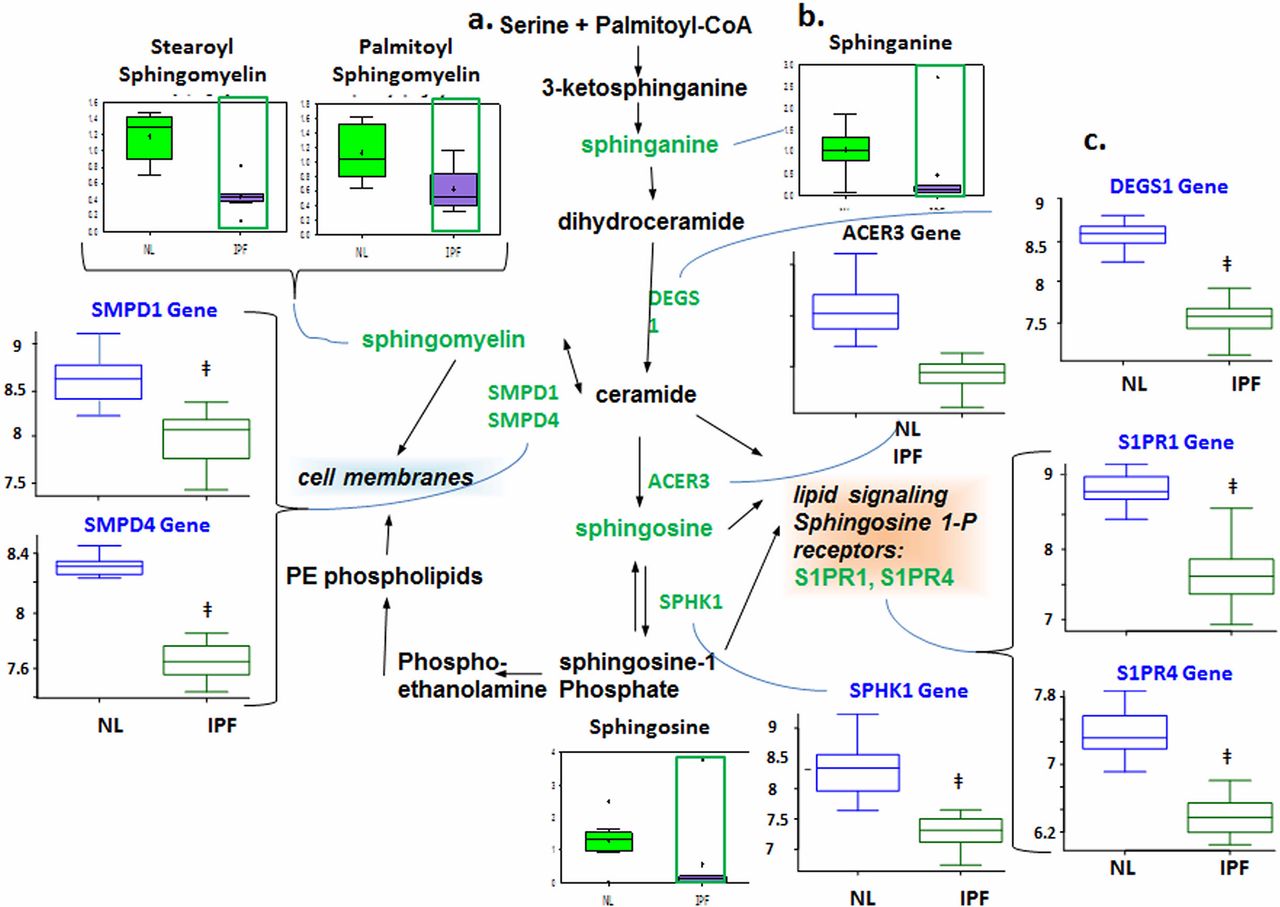
Downregulated sphingolipid metabolism in idiopathic pulmonary fibrosis (IPF) lungs. (A) Data for metabolic intermediates in the normal lung (NL) are shown in green boxes and data for IPF are represented in purple. Quantities are in arbitrary units specific to the internal standards for each quantified metabolite and normalised to protein concentration (n=8 for each box). Samples of patients with IPF exhibited lower levels of sphinganine, sphingosine and sphingomyelin. (B) The classical sphingolipid is shown. (C) Analysis of gene expression for key enzymes of the sphingolipids metabolism; data for control lung are shown in open blue boxes and data for IPF lung are represented in open green boxes (y-axis label of metabolic changes is shown as counts×106). The genes encoding sphingosine 1-phosphate (S1P) metabolic pathway showed downregulated sphingomyelinase (SMPD1 and SMPD4) as well as downregulated delta(4)-desaturase, sphingolipid 1 (DEGS1), suggesting a reduced ceramide production, and downregulated ACER3 (alkaline ceramidase 3) indicates reduced sphingosine and S1P production. Genes encoding DEGS1 (p=1.25e−10), ACER3 (p=2.08e−7), SMPD1 (p=2e−6) and SMPD4 (p=6.67e−14) were significantly changed in IPF lungs compared with normal (y-axis label for gene expression encoding enzymes is shown as fold changes of relative RNA levels of enzymes).
Increased arginine metabolism
The metabolites of arginine are involved in multiple pathways. Creatine participates in ATP production,10 whereas ornithine can be converted to putrescine and spermidine for cell proliferation.11 Ornithine can also be converted to proline and hydroxyproline for collagen formation for fibrosis.12 We found increased levels of creatine, putrescine, spermidine, 4-hydroxyproline and the proline—hydroxyproline dipeptide in patients with IPF compared with normal (figure 2). Interestingly, the levels of arginine itself were not changed. In addition, we found decreased levels of fumarate and aspartate, which are fed into the tricarboxylic acid (TCA) cycle, thereby implicating arginine metabolism as an anaplerotic pathway.

Arginine metabolism is increased in the idiopathic pulmonary fibrosis (IPF) lung. (A) The classical arginine metabolic pathway. (B) In all graphs, the metabolic data for control lung (NL) are shown in green boxes, and data for IPF lung are represented in purple boxes. Quantities are in arbitrary units specific to the internal standards for each quantified metabolite and normalised to protein concentration (n=8 for each box; y-axis label of metabolic changes is shown as counts×106). We found significantly increased polyamines putrescine and spermidine as well as increased 4-hydroxyproline and creatine (p<0.05, increase highlighted in red open box and decrease in green open box). Increased polyamine levels indicate increased cell proliferation, and creatine may provide some of the energy required. Increased 4-hydroxyproline is a marker of fibrosis. Decreased aspartate and fumarate may indicate that arginine metabolism is shuttling intermediates away from the tricarboxylic acid cycle. 5-MTA, 5′-methylthioadenosine; ECM, extracellular matrix; OAT,Ornithine aminotransferase.
Altered energy metabolism in IPF lungs
Energy metabolism is fundamental for any active cell, which implicates its various pathways in the lung structural remodelling process of IPF. We found significant changes in glucose, fatty acid and TCA cycle metabolic pathways in IPF lungs compared with control.
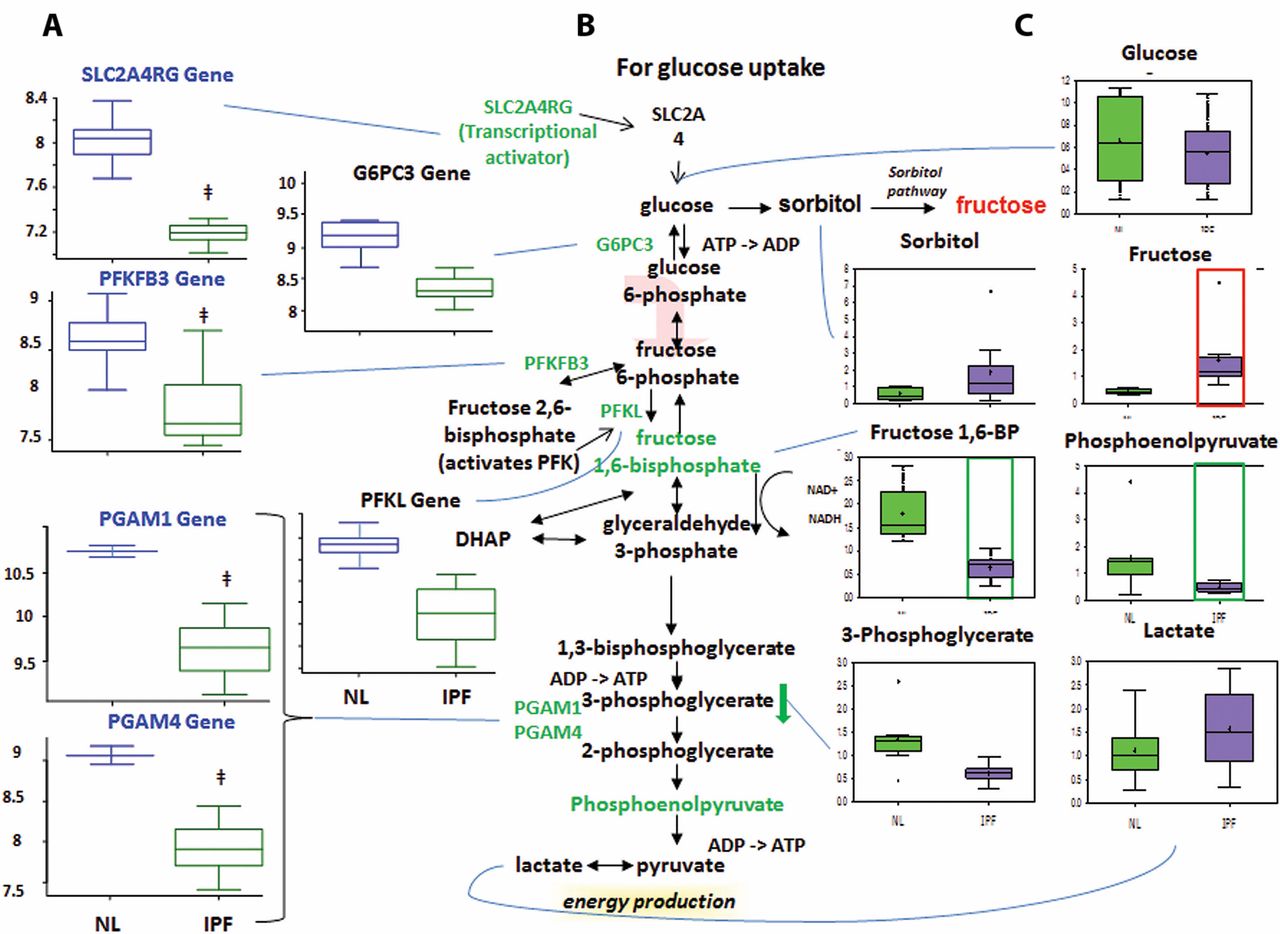
Altered glucose metabolism
Glycolysis generates ATP and converts glucose into pyruvate and then acetyl coenzyme A, which then enters the mitochondria to participate in the TCA cycle.13 Glucose can also be converted to fructose via the sorbitol pathway, and the fructose can then be phosphorylated into fructose 6-phosphate and enter glycolysis.
Decreased glycolysis, altered gluconeogenesis
We found no changes in the early-stage glycolysis metabolites glucose and fructose 6-phosphate (fructose 6-P) (figure 3). However, we did find significantly decreased levels of later-stage glycolysis metabolites fructose 1,6-bisphosphate (fructose 1,6-BP) and phosphoenolpyruvate in IPF lungs compared with normal. This suggests altered glycolysis in IPF lungs. Interestingly, we also found increased lactic acid levels in IPF lungs, which may suggest that the products of glycolysis are being shuttled towards lactic acid production.
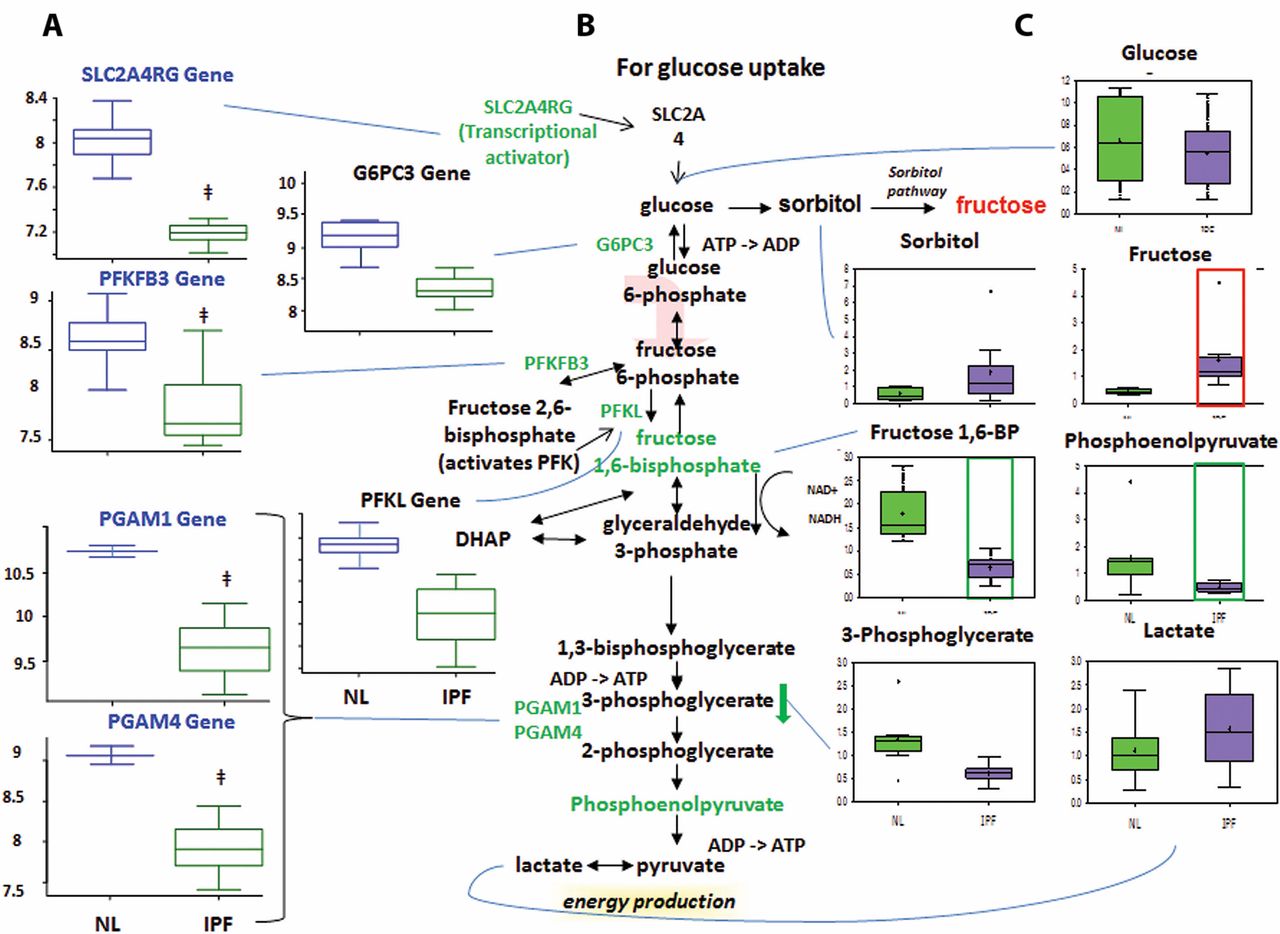
Glycolysis is downregulated in idiopathic pulmonary fibrosis (IPF) lungs, with possible shuttling of intermediates to the sorbitol and pentose phosphate pathways. (A) Analysis of gene expression for key enzymes of glycolysis. Gene-encoding enzymes, including the genes 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), phosphofructokinase (PFK), phosphoglycerate mutase 1 (PGAM1), PGAM4, glucose-6-phosphatase catalytic subunit 3 (G6PC3) and SLC2A4RG, were significantly downregulated in IPF compared with normal suggesting a decreased rate of glycolysis (p<0.05). (B) The classical glycolysis/pentose/energy pathways are shown. (C) Data for metabolic intermediates in the normal lung (NL) are shown in green boxes, and data for IPF are represented in purple boxes. Levels of glycolytic intermediates fructose 1,6-bisphosphate (fructose 1,6-BP) and phosphoenolpyruate were significantly decreased in IPF, suggesting decreased glycolysis. Fructose levels were significantly increased, whereas sorbitol levels showed an upwards trend in IPF, possibly indicating shuttling of glycolytic intermediates towards the sorbitol pathway. The y-axis label of metabolic changes is shown as counts×106 and the y-axis label for gene expression encoding enzymes as fold changes of relative RNA levels of enzymes. DHAP, dihydroxyacetone phosphate.
The microarray data showed decreased expression of solute carrier family 2 (facilitated glucose transporter) (figure 3), member 4 (SLC2A4R) in IPF lungs compared with normal. SLC2A4R transcriptionally activates the inducible glucose transporter SLC2A4 responsible for cellular glucose uptake. This is consistent with our finding that glucose levels were not increased. Importantly, we found decreased expression of phosphofructokinase (PFK), one of the rate-limiting enzymes in glycolysis14 which phosphorylates fructose 6-P to fructose 1,6-BP. This may account for the decrease in fructose 1,6-BP in IPF lungs. We also found decreased levels of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), which interconverts fructose 6-P and fructose 2,6-bisphosphate (fructose 2,6-BP). Since fructose 2,6-BP potently activates PFK,15 16 it is possible that the decreased PFKFB3 expression accounts for the decreased PFKL levels. Taken together, the reduced metabolite levels and the reduced expression of mid-stage to late-stage glycolytic enzymes point to reduced glycolysis in IPF lungs. In addition, we found a reduced levels of glucose-6-phosphatase catalytic subunit-3 (G6PC3) in IPF lung, which converts glucose 6-phosphate into glucose in the final step of gluconeogenesis (figure 3). However, glucose levels were not changed, perhaps because another pathway is compensating for glucose levels or because G6PC3 changes were not sufficient to affect glucose. Therefore, while we found altered levels of genes and metabolites for gluconeogenesis in IPF lungs, it remains inconclusive whether this pathway was upregulated or downregulated.
Sorbitol and pentose phosphate pathways
The sorbitol pathway converts glucose to sorbitol and then fructose, which can be phosphorylated into fructose 6-P and enter glycolysis (Supplementary figure 1). The pentose phosphate pathway converts glucose 6-phosphate into fructose 6-P and glyceraldehyde 3-phosphate for glycolysis. For the sorbitol pathway, our results showed increased fructose levels in IPF lungs compared with normal (Supplementary figure 1) but no change in glucose or sorbitol levels. It remains inconclusive whether the sorbitol pathway was altered, as the increased fructose could have come from other pathways.
Altered fatty acid metabolism
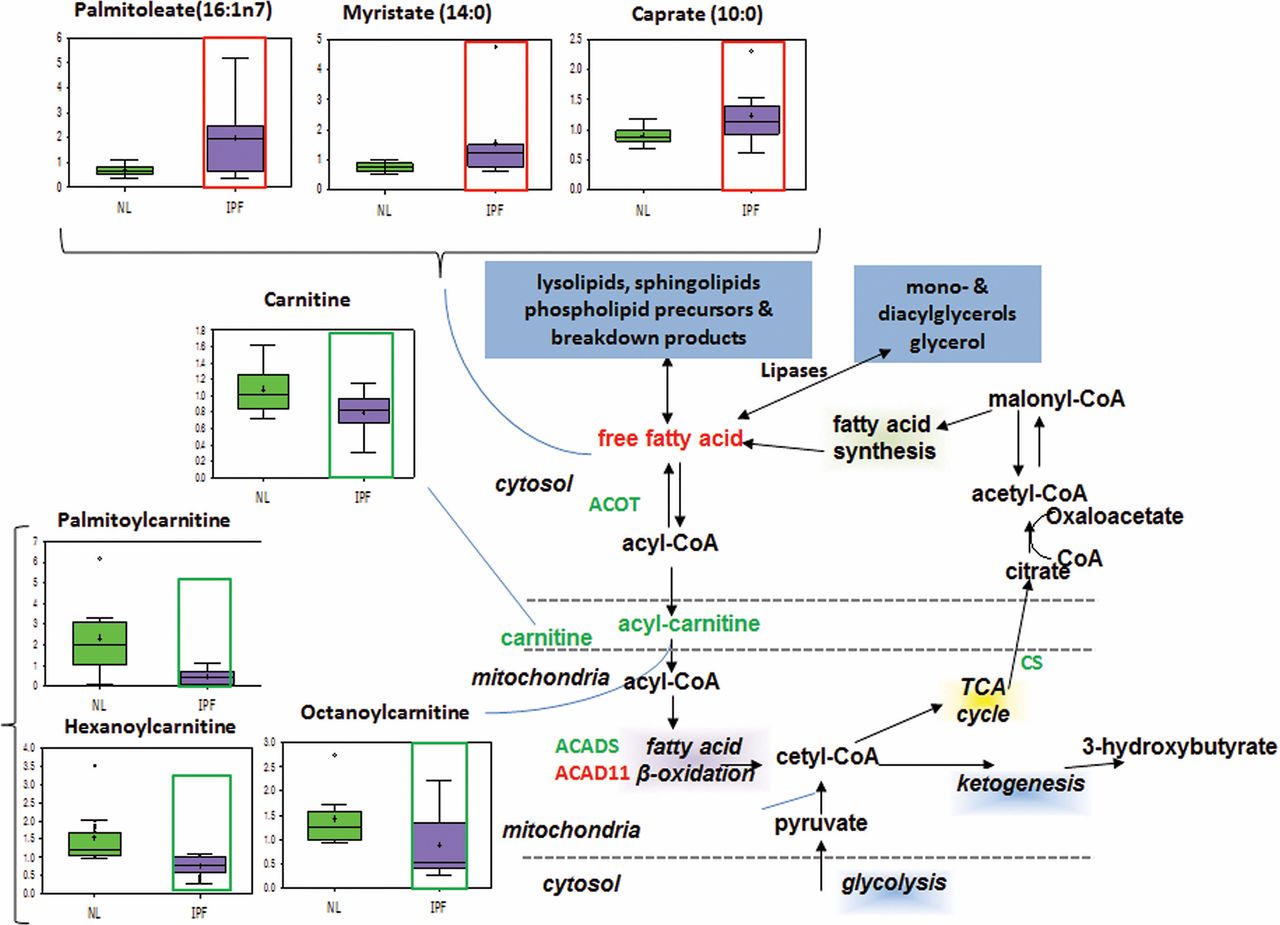
Carnitine transport and mitochondrial beta-oxidation
Fatty acid beta-oxidation occurs primarily in the mitochondria and is important for generating ATP and Nicotinamide adenine dinucleotide (NADH) for energy production.17 It also generates acetyl coenzyme A which participates in the TCA cycle, ultimately leading to ATP synthesis. To enter the mitochondria, fatty acids must be attached to coenzyme A and shuttled into the mitochondria via an acylcarnitine intermediate. Our results showed significantly increased levels of long-chain and medium-chain fatty acids caproate, caprylate, myristate and palmitoleate in IPF lungs compared with normal (Supplementary figure 2), and we also found significantly decreased levels of medium-chain acylcarnitines hexanoylcarnitine, octanoylcarnitine, palmitoylcarnitine and succinylcarnitine (figure 4). The carnitine synthesis pathway produces carnitine and glycine, both of which were found to be decreased in the IPF lung. The data from metabolite analysis suggest that mitochondrial transport is impaired and fatty acids are accumulating in IPF lungs. Since mitochondrial fatty acid import is a vital prerequisite for beta-oxidation,17 we have reason to believe that mitochondrial beta-oxidation is downregulated in the IPF lung.

Mitochondrial transport. In all graphs, the metabolic data for normal lung (NL) are shown in green boxes, and the data for idiopathic pulmonary fibrosis (IPF) lung are represented in purple boxes. Significant increase of fatty acids palmitoleate, caproate and myristate and decrease of acylcarnitines and carnitine shuttle (palmitoylcarnitine, hexanoylcarnitine and octanoylcarnitine) were found in IPF lung compared with normal (p<0.05, increase highlighted in red open box and decrease in green open box). The y-axis label of metabolic changes is shown as counts×106.
In IPF lungs, we found decreased expression of acyl coenzyme A thioesterases 1, 2, 8, 13 (ACOT 1, 2, 8, 13) (Supplementary figure 3), which are responsible for converting fatty acyl coenzyme A into free fatty acids. Interestingly, our gene expression analysis appears to conflict with the concurrent increase in free fatty acids. This may indicate that gene transcription changes in response to increased fatty acids accumulation. Alternatively, another fatty acid production pathway may be compensating for the decrease in ACOT, which will require further analysis.
In beta-oxidation, we found increased expression of ACAD11 but decreased acyl coenzyme A dehydrogenase (ACAD), C-2 to C-3 short chain (ACADS) in IPF lungs compared with normal (Supplementary figure 3). The former enzyme metabolises long-chain fatty acyl coenzyme A, whereas the latter metabolises short-chain fatty acyl coenzyme A. However, if the carnitine-mediated fatty acid mitochondrial transport is indeed impaired, upregulation of ACAD enzymes would not increase beta-oxidation.
TCA cycle in IPF lungs
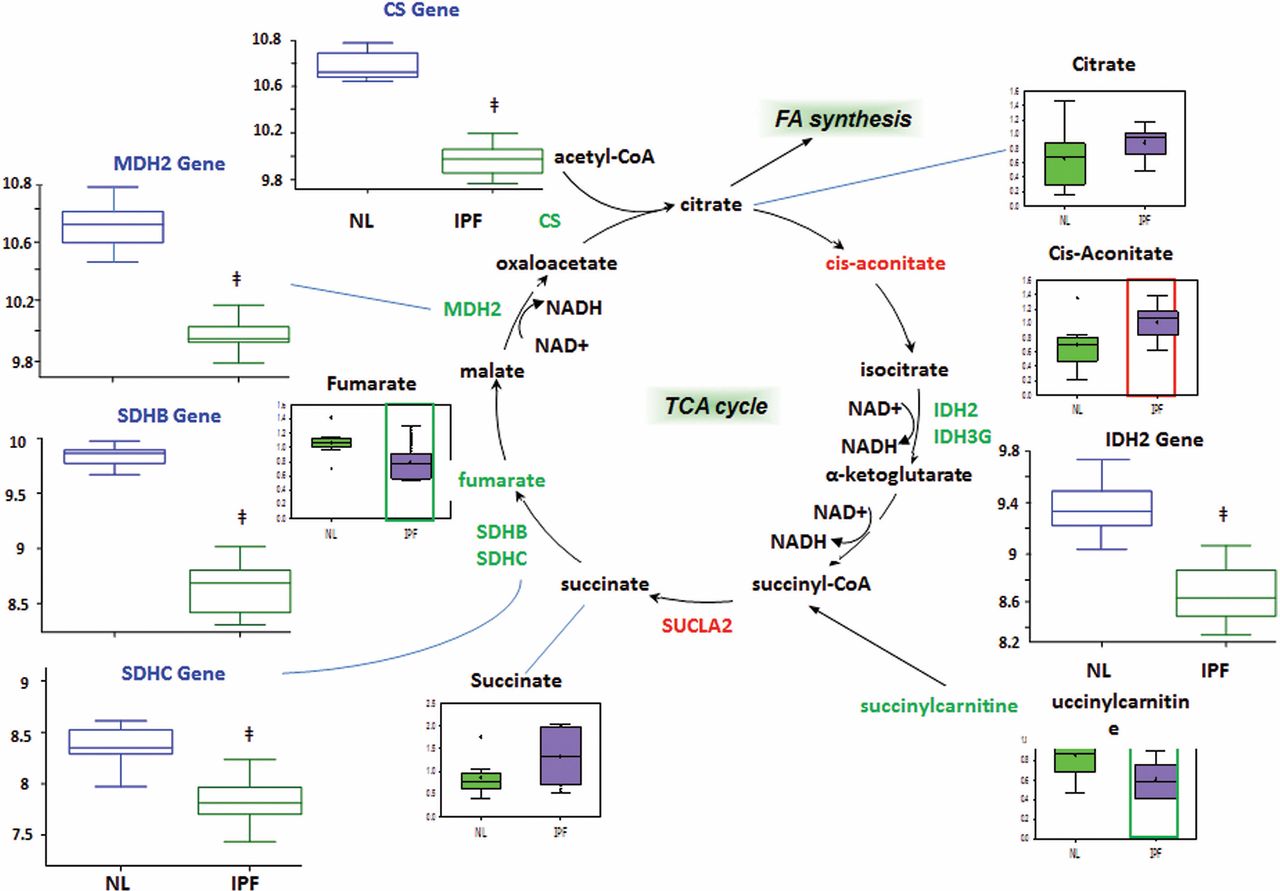
The TCA cycle accepts acetyl coenzyme A generated from beta-oxidation and glycolysis (after pyruvate dehydrogenase converts pyruvate to acetyl coenzyme A) and generates flavin adenine dinucleotide (FADH2) and NADH for the electron transport chain. Therefore, it is an important pathway for cellular energy production. We found significant increases in the enzyme succinyl coenzyme A synthetase (SUCLA2) and the metabolite cis-aconitate (figure 5). There was an increased trend in the SUCLA2 metabolite succinate. Our results also showed significant decreases in various TCA cycle metabolites fumarate and succinyl carnitine (a precursor for succinyl coenzyme A). In comparison, genes encoding the enzymes isocitrate dehydrogenase (IDH2, IDH3G), succinate dehydrogenase (SDHB, SDHC) and citrate synthase (CS) were significantly reduced. The trend is a decrease in TCA cycle enzymes and metabolites, potentially indicating a downregulated TCA cycle in IPF.

Tricarboxylic acid (TCA) cycle. We found overall decreased TCA cycle metabolites and enzymes in idiopathic pulmonary fibrosis (IPF) lungs. In all graphs, the genes encoding metabolic enzymes for control lung (NL) are shown in blue open boxes and IPF lung in green open boxes, whereas metabolic data for control lung are shown in green boxes and IPF lung are represented in purple boxes. Significant increases in the enzyme succinyl coenzyme A synthetase (SUCLA2) and the metabolite cis-aconitate decreases in isocitrate dehydrogenase (IDH), malate dehydrogenase (MDH), fumarate, succinate dehydrogenase (SDH) and citrate synthase (CS) and accumulation of cis-aconitate are found in IPF compared with control (p<0.05). The y-axis label for gene expression encoding enzymes is shown as fold changes of relative RNA levels of enzymes.
Haem metabolism
Haem is used in forming haem-containing proteins and can also be salvaged from its degradation. In haem degradation, haem oxygenase (HO) converts haem to biliverdin, and biliverdin reductase converts biliverdin to bilirubin for excretion. In IPF lungs, we found decreased haem and biliverdin levels, but increased bilirubin ZZ and bilirubin EE levels compared with control (figure 6). However, we found reduced gene expression of uridine 5′-diphospho-glucuronosyltransferases B3GAT3 (not shown) and significant increase of UGT1A1 (figure 6) in the IPF lung, which converts bilirubin ZZ to bilirubin glucuronide for excretion. Taken together, this may indicate an increased haem degradation pathway that depletes haem and accumulates bilirubin.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Haem metabolism. In all graphs, the genes encoding metabolic enzymes for normal lung (NL) are shown in blue open boxes and data for IPF lung are represented in green open boxes, whereas metabolic data for control lung are shown in green boxes. Significantly decreased haem and biliverdin but increased bilirubin levels were found in IPF compared with control (p<0.05). Significantly increased gene expression of uridine 5′-diphospho-glucuronosyltransferase 1 (UGT1A1) was found in IPF lung (p<0.05). The y-axis label of metabolic changes is shown as counts×106 and the y-axis label for gene expression encoding enzymes as fold changes of relative RNA levels of enzymes. ROS, reactive oxygen species.
Glutamine/aspartate metabolism
Glutamate, glutamine and aspartate are involved in several reactions that produce TCA cycle intermediates. Glutamate is converted to the TCA cycle intermediate alpha-ketoglutarate, whereas aspartate is converted to the TCA cycle intermediate oxaloacetate in a reaction that simultaneously converts alpha-ketoglutarate back to glutamate (Supplementary figure 4). We found decreased aspartate levels and increased glutamate and glutamine levels in IPF compared with normal. We also found decreased levels of glutamate dehydrogenase 1 (GLUD1), which interconverts glutamate and alpha-ketoglutarate. These data suggest altered levels of these anaplerotic reactions, which could potentially affect the TCA cycle.
Glutamate can also be converted to glutathione (GSH), which is an antioxidant and has been implicated in pulmonary fibrosis.18 We found increased GSH and decreased levels of its oxidised counterpart glutathione disulfide (GSSG) in IPF lungs compared with normal, which may reflect the body’s attempt to combat oxidative stress in IPF.
Discussion
This study was conducted to analyse the biochemical and gene expression profiles of lung tissue samples in patients with IPF compared with a control group to identify metabolic and genetic biomarkers that may be targeted for the diagnosis and treatment of IPF. We believe that this is the first report that simultaneously and systemically measured the changes of metabolites involving seven metabolic pathways in human advantage of IPF lungs. We have shown various alterations in signalling pathways of sphingolipids, arginine, energy metabolism, haem and glutamate/aspartate metabolism, in patients with IPF compared with control. These pathways, when dysregulated, may underlie IPF pathogenesis.
Our results show downregulated SMPD1, SMPD4 and DEGS1, which point to disrupted ceramide production, whereas downregulated ACER3 and SPHK1 genes indicate disrupted S1P production. This suggests a change in the balance between levels of ceramide and S1P (the so-called ceramide/S1P rheostat), which has been shown to affect cell proliferation19 (Supplementary figure 5). Therefore, a change in the ceramide/S1P rheostat, as seen in our findings, indicates alterations in cell proliferation signalling, possibly for lung structural remodelling. Furthermore, S1P has been implicated in TGF-beta signalling.20 21 Extracellular S1P activates the Rho signalling cascade, which has been shown to cross-activate the connective tissue growth factor (CTGF).20 CTGF has been shown bind TGF-beta and is believed to be the downstream mediator of TGF-beta-induced fibrosis.22 However, intracellular S1P inhibits CTGF expression in vitro.23 This indicates that extracellular S1P enhances fibrosis through cross-activation of the TGF-beta signalling pathway whereas intracellular S1P inhibits it. Previously, Milara et al 24 found increased S1P in human IPF bronchoalveolar lavage (BAL) fluid and increased SPHK1 messenger RNA (mRNA) in human IPF BAL alveolar macrophages, which likely measured extracellular S1P. Our findings of reduced S1P as well as other sphingolipid metabolites are from the lung tissue. Also, we measured SPHK1 mRNA levels, whereas Milara et al measured SPHK1 protein levels in IPF lung tissue, this may account for the discrepancies in results. It would be interesting to confirm whether intracellular S1P is reduced in IPF lungs.
Our group found increases in arginine metabolites creatine, hydroxyproline and the putrescine and spermidine. Other groups have shown increased arginase expression or activity in either human IPF lung tissue or in bleomycin-induced mouse lung fibrosis models.25 26 Arginase converts arginine into ornithine, which is the precursor for proline–hydroxyproline and the polyamines putrescine and spermidine. Therefore, increases in these metabolites can be explained by upregulated arginase. Creatine is involved in energy metabolism. Creatine kinase transfers a phosphate group between ADP and creatine10 to generate ATP. Therefore, increased creatine in our results may be indicative of an upregulation of alternative ATP synthesis pathways to provide energy for lung structural remodelling.
Hydroxyproline is an important component of collagen for the extracellular matrix (ECM). Increased hydroxyproline levels in IPF are in accord with the increased ECM deposition in this disease. Polyamines have been shown to be involved in cell proliferation,11 27 and their depletion has been shown to arrest cell growth in HeLa cells by inhibiting protein synthesis.28 Therefore, increased polyamine levels in IPF lungs suggest increased cell proliferation. It is also interesting to note that fumarate and aspartate levels were found decreased. Since these metabolites also feed into the TCA cycle, increased arginine metabolism may be shuttling intermediates away from the TCA cycle and into ECM deposition and cell proliferation, possibly to support lung structural remodelling.
We found disrupted glycolysis in the IPF lung, with possible increases in the sorbitol and pentose phosphate pathways. Decreased glycolysis is in agreement with findings from Korfei et al,29 who found that GAPDH, PGK, phosphoglycerate mutase 1, TPI and LDHA were increased, whereas PKM, ENOA and FBP1 were decreased. While there was no comment on upregulated or downregulated glycolysis, it can be inferred that since PKM is a rate-limiting enzyme in glycolysis, their findings support reduced late-stage glycolysis, which is in agreement with our results. Discrepancies between our results may be due to other factors during translation or protein synthesis. We also found possible upregulation of the sorbitol and pentose phosphate pathways in IPF, hinting at the shuttling of glycolytic metabolites towards these pathways. Interestingly, in the pentose phosphate pathway, we found increased ribulose 5-phosphate, xylulose 5-phosphate and ribose, although the results were not significant. Korfei et al’s proteomic analysis also found decreased transketolase (TKT), which supports this idea. TKT metabolises xylulose 5-phosphate and ribose 5-phosphate, so its decrease would allow for the accumulation of ribulose 5-phosphate and xylulose 5-phosphate. Ribose is a precursor for nucleotide synthesis, and increased nucleotide synthesis may support cell proliferation and thereby lung structural remodelling in IPF.
We found accumulation of free fatty acids and reduced carnitine shuttle. Since fatty acids, especially longer chain fatty acids, require the carnitine shuttle to enter the mitochondria, this suggests reduced mitochondrial beta-oxidation in IPF. Furthermore, abnormal activity of very long-chain acyl-CoA dehydrogenase (VLCAD), an enzyme involved in the beta-oxidation of very long-chain fatty acids, has been found in human IPF lung fibroblasts.30 However, since mitochondrial beta-oxidation of long-chain fatty acids is dependent on carnitine-mediated transport, a decrease in the carnitine-mediated transport would alter beta-oxidation, regardless of the activity of beta-oxidation enzymes. Overall, glycolysis, mitochondrial beta-oxidation and glucose oxidation via the TCA cycle are major cellular energy pathways. Their downregulation suggests that alternative energy pathways are upregulated to support cell growth and lung structural remodelling. Upregulated creatine metabolism may be an example of one such pathway.
Our findings of reduced haem and biliverdin levels in IPF, together with increased bilirubin levels, suggest increased haem degradation to produce bilirubin. Previously, Nakamura et al 31 found that increasing HO-1 levels reduces TGF-beta-stimulated collagen production in vitro via carbon monoxide (CO) rather than bilirubin, whereas Atzori et al 32 found that increased HO-1 attenuates fibrosis in bleomycin-induced mouse lung fibrosis models. Our findings of decreased haem levels may therefore result in reduced CO. Since CO can inhibit TGF-beta-induced fibrogenesis, reduced haem and therefore CO levels may contribute to fibrosis. Oxidative stress has been proposed to play a role in IPF pathogenesis, possibly by inducing alveolar epithelial injury.33 Indeed, lung epithelial cells of patients with interstitial pneumonia have been found to contain higher levels of oxidants than normal tissue.34 Bilirubin is an antioxidant and has been shown to attenuate bleomycin-induced rat pulmonary fibrosis.35 Therefore, our findings of increased bilirubin may be due to attempting to reduce oxidative stress in IPF.
Our group found increased glutamine and glutamate and decreased aspartate levels in IPF lungs, although the changes were not significant. Since this pathway is an anaplerotic pathway to generate intermediates for the TCA cycle changes in this pathways may account for the reduced TCA cycle metabolites in IPF. We also found increased levels of the antioxidant glutathione (GSH, the reduced form) and decreased levels of its oxidised counterpart GSSG in IPF lungs. This supports previous findings that bleomycin activates the oxidative stress response in the bleomycin-induced mouse lung fibrosis model and upregulates GSH. Liu et al 36 also found that GSH levels were initially suppressed by overexpression of TGF-beta in mouse lungs, but later returned to normal, possibly implicating GSH in a response to fibrosis. Our findings may therefore reflect the body’s attempt to generate more GSH as part of the oxidative stress response in IPF.2
Study limitations
The number of subjects in the current study is relatively limited. However, our sample size is equivalent to similar studies in this field as it is very difficult to obtain fresh human IPF lungs. Although the tissues for all studies were collected within 2 years, this short period may still be a limitation to obtaining more tissue for both metabolomics and transcription study.
The results of this study support a greater understanding of the metabolic basis of pathogenesis of IPF formation. From a clinical perspective, this may translate to new developments of methods to assess IPF metabolism (as a predictor of prognosis or as a way to assess the efficacy of IPF targeted therapy).
Finally, understanding the metabolic pathways in the development of IPF provides the possibility of exploring the effects of metabolic modulation as a potential therapeutic strategy in the treatment or prevention of IPF.
Conclusions
The current study indicates that there is a metabolic shift in the advantage of IPF formation. Those metabolite changes may represent group marker for the prediction of the development of IPF when compared with that of PAH and other lung diseases. Metabolic modulation may be a target for the development of novel therapies in IPF management. The changes of multiple metabolic pathways may associate with clinical observations. Further studies will be performed to determine whether metabolic imaging has a role in the diagnosis and management of IPF.
Acknowledgments
The authors wish to thank Hana, ZH Yun for her excellent technical support.
References
Footnotes
Contributors Conception and design: YDZ, JG and MdP.
Draft: YDZ, JG, MdP, SA, TKW, SK.
Experiment performance and analysis and interpretation: YDZ, LY, CL, GZ, YY, LW, MH.
Competing interests None declared.
Ethics approval Research protocols approved by the institutional review boards of the University Health Network.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.