Article Text

Abstract
Introduction Chlamydia pneumoniae respiratory tract infection has been implicated in the pathogenesis of reactive airway disease and asthma. Innate cytokine responses that are protective of infection with intracellular pathogens may be impaired in patients with asthma. Tumour necrosis factor alpha (TNF-α) is a cytokine related to functions of monocytes and may inhibit C. pneumoniae infection. We investigated TNF-α responses in C. pneumoniae-infected peripheral blood mononuclear cells (PBMCs) in patients with asthma and non-asthma, and whether ciprofloxacin, azithromycin or doxycycline affects TNF-α responses.
Methods PBMC (1.5×106) from paediatric patients with asthma (n=19) and non-asthmatic controls (n=6) were infected or mock infected for 1 hour with or without C. pneumoniae AR-39 at a multiplicity of infection=0.1, and cultured+ciprofloxacin, azithromycin or doxycycline (0.1 ug/mL) for 48 hours. TNF-α levels were measured in supernatants by ELISA.
Results When PBMC from patients with asthma were infected with C. pneumoniae, levels of TNF-α were significantly lower than in subjects without asthma (48 hours) (5.5±5.6, 38.4±53.7; p=0.0113). However, baseline responses (no infection with C. pneumoniae) were similar in asthma and non-asthma (1.0±1.7, 1.1±1.2; p=0.89). When PBMC frompatiens with asthma were infected with C. pneumoniae+ciprofloxacin, azithromycin or doxycycline, TNF-α levels increased (25%–45%); this affect was not observed in PBMC from patients without asthma.
Conclusions We identified differences in the quantity of TNF-α produced by C. pneumoniae-infected PBMC in asthma compared with non-asthma.
- asthma
- bacterial infection
- cytokine biology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Using our Chlamydia pneumoniae model, can we determine whether innate cytokine responses (eg, tumour necrosis factor alpha (TNF-α)) that are protective of infection with intracellular pathogens (eg, C. pneumoniae) impaired in children with asthma?
Higher levels of TNF-α are produced by C. pneumoniae-infected peripheral blood mononuclear cell from patients without asthma compared with patients with asthma, but TNF-α responses increased after addition of ciprofloxacin, azithromycin or doxycycline in asthma.
These data suggest that TNF-α cytokine responses may be impaired in patients with asthma, and treatment with antibiotics modulates TNF-α responses in asthma.
Introduction
Chlamydia pneumoniae is a Gram-negative obligate intracellular bacterium that causes respiratory infection in adults and children.1 2 Studies have shown that C. pneumoniae can cause prolonged respiratory infection in subjects with asthma and without asthma3–5 and has been implicated in asthma exacerbation.2 C. pneumoniae infection can activate monocytes/macrophages, dendritic cells, endothelial cells and epithelial cells to produce cytokines that may contribute to the pathology observed in asthma2 and trigger both innate and adaptive immune responses.6 Human monocytic cells are triggered to release proinflammatory cytokines including tumour necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-6 and IL-8.6 7
Prior literature has reported in a mouse model that host immune responses to C. pneumoniae occur in two stages: an early response requiring interferon (IFN)-γ (from innate immune cells) to limit the growth of the intracellular bacteria, which plays a central role in the innate control of this infection, and a later adaptive immune response that includes CD4+ and CD8+ T cells in bacterial clearance and protection.8 9 The primary immune response is aimed to clear the primary infection from the host and provide protection against reinfection with the same pathogen.8 9
It has been well established that cytokines, including IFN-γ, can restrict growth of intracellular bacterial pathogens and can activate host cell immune responses to infection, including C. pneumoniae.10 TNF-α is a cytokine that can also regulate inflammation and host defence against infectious agents including C. pneumoniae.10 Shemer-Avai et al found in HEp-2 cell cultures that TNF-α was an anti-inflammatory mediator and inhibited intracellular replication of C. trachomatis.11 They also found synergism between IFN-γ and TNF-α in suppression of chlamydial multiplication.11 Previous studies in our laboratory reported that C. pneumoniae modulates IFN-γ responses in peripheral blood mononuclear cell (PBMC) from paediatric patients with asthma, even in absence of active infection.12 Recent studies in our laboratory demonstrated that C. pneumoniae-induced IFN-γ production was more prevalent in paediatric patients with asthma compared with non-asthmatic controls and that C. pneumoniae infection led to a significant increase of IFN-γ levels in supernatants of PBMC from patients with asthma, compared with patients without asthma.13 Although the importance of IFN-γ in bacterial infections is well established, the role of TNF-α during the course of bacterial infections is less defined.
Treatment of infections due to C. pneumoniae with antibiotics (macrolides, tetracyclines and quinolones) may have beneficial effects in patients with asthma due to eradication of acute C. pneumonaie infection2 14 and potential pleiotropic anti-inflammatory properties in addition to antichlamydial activity.14 Quinolones (levofloxacin and moxifloxacin), azithromycin and doxycycline are antibiotics that are frequently used for the treatment of C. pneumoniae respiratory infections.14 Ciprofloxacin is significantly less active than moxifloxacin and levofloxacin and is not used to treat C. pneumoniae respiratory infections in patients14; however, ciprofloxacin has been used in the in vitro setting to test drug class, as well as anti-inflammatory activity independent of antimicrobial activity.15
In this study, we investigated the ability of C. pneumoniae-infected PBMC from patients with asthma and without asthma to produce TNF-α and whether ciprofloxacin, azithromycin or doxycycline affect TNF-α responses because this cytokine plays an important role in antichlamydial activity. This area of focus is of interest from an immunological and clinical perspective; these responses may contribute to susceptibility in persistent C. pneumoniae infections and thus provide a model for emergence of persistent infection. In addition, these findings may reveal new insights into TNF-α’s contribution to protective as well as pathogenic inflammatory responses in C. pneumoniae infection. Specifically, treatment with certain antichlamydial antibiotics may eradicate infection with C. pneumoniae because of the significant effect on TNF-α production by host cells.
Methods
Study population and study participants
Paediatric patients with asthma (male and female; 7–17 years old) were recruited from the outpatient department at SUNY Downstate Medical Center (Brooklyn, New York). Inclusion criteria included either a physician’s diagnosis of stable asthma or current clinically defined persistent asthma symptoms16 or both. Exclusion criteria included history of chronic immunosuppressive or autoimmune disease, HIV infection, cancer, recent use of systemic corticosteroids (<30 days), antibiotic use or immunotherapy and incomplete follow-up. The control subjects (patients without asthma; male and female; 9–16 years old) were defined based on clinical criteria17 and did not have asthma. Specimens were collected at the time of diagnosis, follow-up or referral to our clinic. All procedures were followed in accordance with institutional guidelines involving human subjects. Participants and/or legal guardians provided written informed consent for this study.
Detection of C. pneumoniae-specific IgG antibodies
Using the microfluorescence (MIF) test (AniLabsystems; Vantaa, Finland), we measured C. pneumoniae-specific IgG antibodies, as previously described.18
Preparation of C. pneumoniae
C. pneumoniae AR-39 (ATCC 53592; Manassas, Virginia, USA) was propagated as previously described.19 HEp-2 cell (ATCC CCL-23) monolayers were inoculated with C. pneumoniae, then grown to high titres (serial passage). C. pneumoniae elementary bodies (EBs) were purified by Urografin (Schering, Berlin, Germany) density gradient centrifugation and resuspended in sucrose phosphate glutamate buffer (comprising 74.62 g/L sucrose (Sigma, St Louis, Missouri, USA), 0.517 g/L KH2PO4 (Sigma), 1.643 g/L K2HPO4 (Sigma) and 0.907 g/L potassium glutamate (Sigma)). In order to determine titres, we infected HEp-2 cells with serial dilutions of EB suspension aliquots, fixing cells at 72 hours postinfection (p.i.), then staining with fluorescein-conjugated murine monoclonal genus-specific antilipopolysaccharide monoclonal Ab (Pathfinder, Bio-Rad, Hercules, California, USA). Inclusions per well were then counted. Aliquots were frozen at −80°C.
Cell culture
PBMC were separated from blood (10 mL) on a Ficoll-Paque (GE Healthcare, Sweden) gradient (density 1.077), as previously described.12 13 The PBMCs were removed from the buffy coat. Cells were washed twice in RPMI 1640 medium (Life Technologies/GIBCO, Grand Island, New York, USA) with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Norcross, Georgia, USA), and resuspended in complete RPMI 1640 (c-RPMI). c-RPMI contained RPMI 1640 Medium HEPES Modification (Sigma) supplemented with 5 mM L-glutamine (Sigma) and 10% FBS (Atlanta Biologicals). Cells were counted on a haemocytometer (Fisher Scientific, Springfield, New Jersey, USA). We evaluated cell viability, as judged by trypan blue (Fisher Scientific) exclusion. For each experimental condition, PBMCs (1.5×106/mL) were cultured in duplicate in a 24-well flat bottom plate (1 mL/well) (Corning Inc; Corning, New York, USA) at 37°C in c-RPMI medium in a humidified 5% CO2 atmosphere for up to 12 days. We determined cell viability at 0, 48 and 240 hours (>98%, 95% and 90%, respectively), in the absence of any infection with C. pneumoniae.
Following a 2-hour incubation to allow adherence, PBMCs were infected with C. pneumoniae (by adding purified EB for 1 hour) or mock-infected and/or stimulated in the presence or absence of ciprofloxacin (0.1) (Sigma), azithromycin (0.1 ug/mL) (Sigma) or doxycycline (0.1 ug/mL) (Sigma) for up to 12 days, as previously described.15 20 All antibiotics were serially diluted (1:1, 1:2, 1:4 and 1:10)20 to determine optimal dose and chose doses that gave optimal suppression (for the purpose of cytokine production cytokine assays (TNF-α). Supernatants collected from above cultures were assayed for TNF-α (ELISA).
In vitro infection with C. pneumoniae and treatment with antibiotics
PBMCs were infected with C. pneumoniae by adding purified EB for 1 hour or treated once with concentrations of 0.1 or 1.0 ug/mL of ciprofloxacin, azithromycin or doxycycline for up to 12 days p.i. at 37°C in c-RPMI in a humidified 5% CO2 atmosphere. The multiplicity of infection (MOI; 0.1) and time points (48 hours and 10 days p.i. for cytokines)20 used for analysis were selected by kinetic and dose response studies (using MOI of 0.01–10) for optimisation of the assay, which revealed peak concentrations and clear distinctive profiles for the respective outcome variables at these time points. In order to confirm and quantify infection with C. pneumoniae, adherent cells were stained with a fluorescein-conjugated murine monoclonal genus-specific antilipopolysaccharide antibody (Becton-Dickinson (BD) Biosciences, San Jose, California, USA), at 72 hours p.i. Controls used in infection experiments were: identical volumes of heat-inactivated purified C. pneumoniae 20 and identical volumes of HEp-2 cell cultures not containing any bacteria processed the same way as the purified C. pneumoniae 19 based on dose-response experiments.
Cytokine determination (TNF-α): ELISA
For the in vitro quantitative determination of human cytokine TNF-α: in cell culture supernatants, solid-phase sandwich ELISA assays were performed using LEGEND MAX Human TNF-α ELISA kit (Bio Legend, San Diego, California, USA) ELISA test kits, according to the manufacturer’s recommended procedure.
Cell culture supernatants were collected at either 48 hours or 10 days p.i. by centrifugation, and samples were stored at −80°C until analysis. All specimens were analysed in duplicate, and standard curves were determined. Plates were read using an automated microplate reader (Model ELx800; Bio-Tek Instruments, Winooski, Vermont, USA), with a 450 nm measurement filter. Detection limit for TNF-α assay was <3.5 pg/mL.
Quantitative real-time PCR (qPCR) (swabs and cultures)
Extraction of bacterial DNA from nasopharyngeal swab specimens21 and PBMCs were performed using a QIAAmp DNA Mini-Kit (Quiagen, Valencia, California, USA) according to manufacturer’s recommendations. For PBMC cultures, supernatants with adherent and non-adherent cells were collected and bacterial DNA extracted. Specimens were tested by real-time PCR for the presence and quantification of C. pneumoniae and Mycoplasma pneumoniae DNA according to Apfalter et al 21 and Waring et al,22 respectively, using TAQMan technology-based quantitative real-time PCR on a Light Cycler 2.0 platform (software version 4.0, Roche Diagnostics, Indianapolis, Indiana, USA).
Statistical analysis
Data are expressed as means with SD unless otherwise indicated.
Student’s t-test and the non-parametric Wilcoxon signed ranks test were used to compare differences in means of normally and non-normally distributed data, respectively. The Pearson correlation test was used to assess the degree of correlation for continuous variables. A two-tailed p value of <0.05 was taken to indicate statistical significance for all comparisons. All data and statistical analyses were performed using SPSS for Windows, V.12.0 software (Chicago, Illinois, USA).
Results
Participant characteristics and demographics
We enrolled 19 paediatric patients with asthma (11 males, 8 females) and six non-asthmatic controls (4 males, 2 females). The mean age of the patients with asthma and without asthma was 12±3 years (range: 7–17) and 13±3 (range: 9–16), respectively. None of the healthy subjects had a history of asthma or allergic rhinoconjunctivitis.
All the children with asthma were classified as having moderate persistent asthma and were treated with inhaled corticosteroids (ICS). The ICS used were either fluticasone HFA (176 µg, 220 µg or 440 µg), fluticasone/salmeterol HFA (180 µg), fluticasone/salmeterol DPI (500 µg or 1000 µg), beclomethasone HFA (160 µg) or budesonide suspension (0.5 mg). Total serum IgE levels in asthma were 157±121 IU/mL and <100 IU/mL in non-asthma.
Serological and nucleic acid amplification testing for C. pneumoniae
All (100%) of the patients with asthma had C. pneumoniae-specific IgG MIF titres >1:16 with a median titre of 1:32. All patients tested negative for C. pneumoniae and M. pneumoniae, as determined by qPCR (nasopharyngeal swabs). In contrast, 50% of the control subjects had C. pneumoniae IgG titres >1:16 (median titre 1:32).
C. pneumoniae induced TNF-α responses in asthma compared with non-asthma
When PBMC from patients with asthma were infected with C. pneumoniae, levels of TNF-α were significantly lower than in subjects without asthma (48 hours) (5.5±5.6, 38.4±53.7; p=0.0113) (figure 1). However, baseline responses (no infection with C. pneumoniae) were similar in asthma and non-asthma (1.0±1.7, 1.1±1.2; p=0.89) (figure 1). In addition, there were no differences observed when comparing asthmatic subjects currently using ICS compared with those that did not use them (p=0.364).

Chlamydia pneumoniae -induced TNF-α responses in asthma compared with non-asthma. PBMC (1.5×106) from patients with asthma (n=19) and subjects without asthma (n=6) were infected with C. pneumoniae (MOI=0.1). Levels of TNF-α were measured from supernatants collected on day 2 of culture (48 hours) (ELISA). Data are expressed as pg/mL (mean±SD). *PBMC+C. pneumoniae with asthma versus PBMC+C. pneumoniae with no asthma. *Two-tailed p value statistically significant. MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cell; TNF-α, tumour necrosis factor alpha.
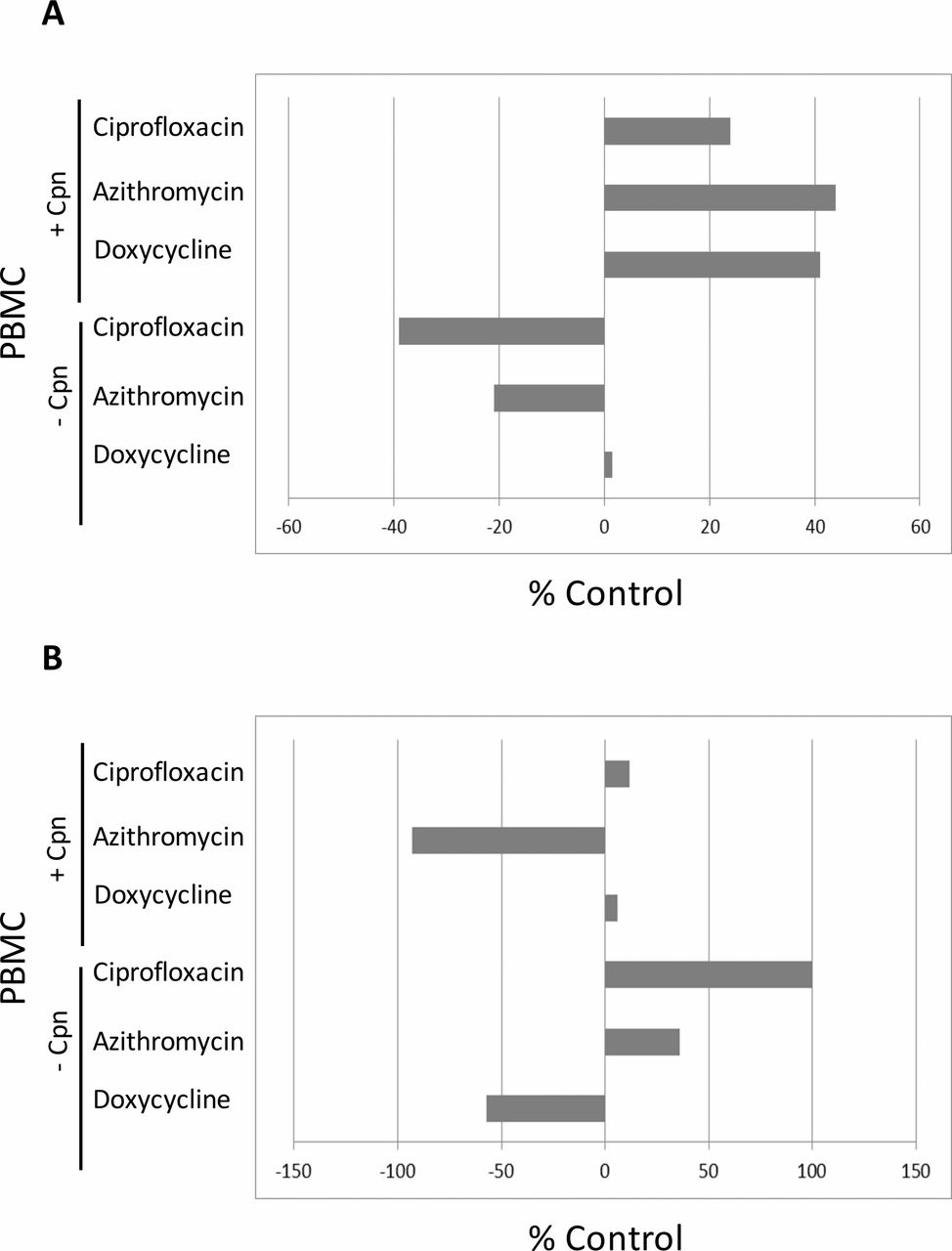
Effect of ciprofloxacin, azithromycin or doxycycline on C. pneumoniae-induced TNF-α responses
In PBMC from patients with asthma infected with C. pneumoniae, TNF-α responses increased after addition of ciprofloxacin (0.1 µg/mL), azithromycin (0.1 µg/mL) or doxycycline (0.1 µg/mL) (25%–45%) at 48 hours (figure 2A). In PBMC from patients with asthma not infected with C. pneumoniae, TNF-α responses did not increase, after addition of ciprofloxacin (0.1 µg/mL), azithromycin (0.1 µg/mL) or doxycycline (0.1 µg/mL) (figure 2A). In PBMC from subjects without asthma infected or not infected with C. pneumoniae, TNF-α responses did not increase after addition of ciprofloxacin (0.1 µg/mL), azithromycin (0.1 µg/mL) or doxycycline (0.1 µg/mL) (figure 2B).
{kind=link}
{kind=link}
Effect of ciprofloxacin, azithromycin or doxycycline on Chlamydia pneumoniae-induced TNF-α responses. PBMC (1.5×106) from patients with asthma (n=19) (figure 2A) and subjects without asthma (n=6) (figure 2B) were infected ±C. pneumoniae (MOI=0.1), and then cultured with ciprofloxacin (0.1 µg/mL), azithromycin (0.1 µg/mL) or doxycycline (0.1 µg/mL). Levels of TNF-α were measured from supernatants collected on day 2 of culture (48 hours) (ELISA). Data are expressed as % control. % control was calculated as follows: % control=1− (TNF-α production in PBMC+treatment/TNF-α production in PBMC without treatment) × 100(%). Cpn, C. pneumoniae; MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cell; TNF-α, tumour necrosis factor alpha.
Discussion
In this study, we examined TNF-α responses in C. pneumoniae-infected PBMC in patients with asthma compared with non-asthma and the effect of ciprofloxacin, azithromycin or doxycycline on the production of TNF-α secreted by C. pneumoniae-infected PBMC. The data revealed that higher levels of TNF-α are produced by C. pneumoniae-infected PBMC from patients without asthma compared with patients with asthma. In addition, we observed that in C. pneumoniae-infected PBMC from patients with asthma, TNF-α responses increased (25%–45%) after addition of ciprofloxacin, azithromycin or doxycycline. Innate cytokine responses that are protective of infection with intracellular pathogens are impaired in patients with asthma.
TNF-α is a pleiotropic cytokine involved in inflammation, apoptosis and host defence.23 TNF-α acts both as proinflammatory mediator (initiating strong inflammatory responses) and as immunosuppressive mediator (inhibiting development of autoimmune disease) and plays a role in tumourigenesis.23 TNF-α maintains immune homeostasis through regulation of inflammatory responses23 and is also involved in allergic disease development (eg, asthma)24; it plays a role in host defence against viral, bacterial, fungal and parasitic pathogens, as well as intracellular bacterial infections, including C. pneumoniae, Mycobacterium tuberculosis and Listeria monocytogenes.23 25 It should be mentioned that C. pneumoniae can stimulate toll like receptor (TLR)-4 expression in rat type II pneumocytes, leading to the production of TNF-α and macrophage inflammatory protein-226; TLR-4 plays an important role in innate pulmonary immune defence mechanism and clearance of C. pneumoniae.26
The main finding in the present study was that higher levels of TNF-α are produced by C. pneumoniae-infected PBMC from patients without asthma compared with patients with asthma. Addition of either ciprofloxacin, azithromycin or doxycycline increased TNF-α responses in C. pneumoniae-infected PBMC from the patients with asthma. It can be speculated that the cytokine responses observed likely reflect the cumulative responses of many cell types. The differences observed between patients with asthma and without asthma suggest that patients with asthma may have altered immune responses, that is, a deficient Th1 immune response (innate immune dysfunction) to bacterial infection, perhaps due to deficient dendritic cell function, thus increasing susceptibility to infections. However, the molecular mechanism for this is unclear. The lower levels of TNF-α in asthma were not likely due to steroid use, according to our observations. However, with less TNF-α, there is more in vitro proliferation of C. pneumoniae 27; this may explain why children with asthma are more likely to have an IFN-γ response to C. pneumoniae (indicating infection). Increasing TNF-α responses, through antibiotic treatment, may limit the proliferation and spread of C. pneumoniae. The clinical significance of an observed increase in the TNF-α response after addition of antibiotic is uncertain but may have important implications for treatment of bacterial pathogens in subjects with asthma.
Our data are in agreement with contrasting observations on the production of cytokines in immune regulatory responses in C. pneumoniae-infected cells.20 Even though our results may seem counterintuitive to research demonstrating that overexpression of TNF-α may lead to harmful inflammation and adverse health outcomes in patients with asthma,28 our study focuses on specific stimulant C. pneumoniae, which has been shown to modulate TNF-α responses28; this may be independent of other TNF-α responses. It should be mentioned that we have not fully characterised the genetic background of our study subjects in terms of TNF gene polymorphisms.
Our findings may indicate that chronic inflammation due to C. pneumoniae infection may affect immune responses differently in individuals without asthma compared with those with asthma. C. pneumoniae infection may negatively affect TNF-α-mediated immune effector pathways in susceptible cells through unknown mechanisms. Induction of chronic inflammatory responses by C. pneumoniae infection may also promote progression of asthma by triggering monocytes/macrophages to secrete TNF-α.
Given the findings reported, it should be mentioned that similar differential baseline production or production in response to other infections may be a common feature to select populations. Thus, it would be feasible to apply similar methodologies to young children with asthma in response to viral (ie, respiratory syncytial virus, influenza virus and human rhinoviruses) or other infections, which are common triggers for exacerbation. If select antibiotics can help enhance cytokine responses to infection in general, our model can potentially provide a rationale for alternate therapies and be used in children with other suspected infections that have a similar type of protective immunity as chlamydial infections.
Netea et al 29 demonstrated that stimulation of healthy human PBMC with sonicates of C. pneumoniae can induce chemokines and proinflammatory cytokines (TNF-α, IL-1, IL-6 and IL-8). In contrast, when we compared cytokine responses between healthy volunteers and subjects with asthma, we found decreased TNF-α production in subjects with asthma. In view of the increased prevalence of C. pneumoniae infection in individuals with asthma, a decreased TNF-α response may be significant for ineffective immune response and clearance. Thus, this may be a possible mechanism that helps explain the clinical finding of why C. pneumoniae-infected paediatric patients with wheezing did not clear the infection, in addition to demonstrating persistent infection (up to 5 months).3
Prior literature has reported that IL-12 and TNF-α are produced early in intracellular bacterial infection.30 Zhan et al 30 demonstrated that TNF-α and IL-12 are both required for resistance to Brucella abortus, which is also an intracellular organism, although their roles are different.
It is possible that there may exist deficient immune responses due to decreased IL-12 secretion in the asthmatic state.31 Hofstra et al 31 reported that in an experimental asthma model in BALB/c mice, IL-12 can contribute to allergic airway disease secretion, resulting in impaired inhibition of Th2 cell differentiation, leading to an exaggerated Th2 responses. IL-12-related genes are important in T cell immunity; cytokine pathways are involved in the onset and chronic state of the disease.31 Previous studies from our laboratory demonstrated that C. pneumoniae infection induces IgE production and modulates IL-12 responses in patients with asthma, which may be caused, in part, by differences in TLR-2 and TLR-4 stimulation.32 Studies from our laboratory have now confirmed that the production of two innate cytokines (IL-1232 and TNF-α) is decreased due to C. pneumoniae infection in asthma compared with healthy volunteers and speculate that there may exist a synergistic mechanism responsible for the observations.
In contrast to the aforementioned studies, previous studies from our laboratory demonstrated that C. pneumoniae-induced IFN-γ production was more prevalent in patients with asthma compared with non-asthmatic controls.12 13 C. pneumoniae infection also led to a significance increase of IFN-γ levels in supernatants of PBMC from patients with asthma.12 13 These results may indicate recent or persistent C. pneumoniae infection in patients with asthma. Kohlhoff et al,33 demonstrated that persistent infection appears to be a critical factor in modulation of host cell apoptosis by C. pneumoniae; induction of apoptosis leading to host cell lysis may help propagate the infection, while inhibition of apoptosis could help protect the organism when it is in the persistent state.33 This is agreement with the current investigation finding of suppressed TNF-α production that may further protect C. pneumoniae in its persistent state in patients with asthma. It can also be speculated that lower TNF-α levels observed in asthma, as well as the fact that a higher percentage of children with asthma have IFN-γ responses,13 which may reflect ineffective Th1 (protective) responses and provide evidence for failure to clear chlamydial infections in this population. The idea of decreased TNF-α responses in asthma may provide an important mechanistic link of how Th1/Th2 pathways can negatively regulate immune responses. It is conceivable that in patients with asthma, there is a predominance of M2 macrophages that secrete lower amounts of TNF-α in response to infectious stimuli.34
In patients with asthma, it is unclear whether the presence of IFN-γ producing C. pneumoniae-infected PBMC, and the levels of IFN-γ produced, represent an effective Th1 response reflecting more frequent infections, or rather a higher prevalence of latent infection due to ineffective clearance. However, other studies in our laboratory demonstrated that C. pneumoniae-infected PBMC from patients with asthma produced increased levels of IL-4 and IFN-γ (IL-10 and IL-12 were low); uninfected PBMC from patients with asthma stimulated with Lactobacillus rhamnosus GG produced undetectable levels of IL-4, while IL-10, IL-12 and IFN-γ levels increased.35 These results suggest that C. pneumoniae infection may promote Th2 responses in C. pneumoniae-infected PBMC from patients with asthma. This may be due to activation of receptors for pathogen-associated molecular patterns.35 There seems to exist a clear difference between the intracellular bacterium Chlamydia and the majority of other bacteria because of differences in their cell wall components and stimulatory properties.
Our study has several limitations that should be kept in mind in the interpretation of the findings, including a small sample size from a single patient population that requires confirmation for generalisation. Serological methods for retrospective confirmation of timing of infection may be difficult, and serology often poorly correlates with both active and past infection. Thus, incidence of primary infection in patients is unknown. In addition, ICS taken by the patients with asthma may have contributed to the observations that TNF-α levels were lower in patients with asthma. However, in our study, we did not find a statistically significant difference between patients with asthma currently using ICS and those that did not use them. Lastly, the target organ of asthma inflammation is the lung, which was not studied directly in the current study. However, given the existing abundance of scientific evidence using PBMC as markers of inflammation, we believe that our model provides relevant information. It would be of interest, in future studies, to compare biomarkers of infection in lung fluids versus blood.
In summary, we found that C. pneumoniae-induced TNF-α responses in PBMC were lower in asthma compared with non-asthma. It could be that innate cytokine responses that are protective of infection with intracellular pathogens are impaired in children with asthma. TNF-α responses increased after treatment with ciprofloxacin, azithromycin or doxycycline.
Acknowledgments
We would like to thank Kevin Norowitz, MD (SUNY Downstate Med Center, Department of Pediatrics) for fruitful discussions.
References
Footnotes
Contributors All authors listed on this manuscript made substantial contributions to the conception (TAS-N, MRH and SK) or design of the work (YMN and DW) or the analysis (KC) and interpretation of the work (RJ). TAS-N and SK wrote and approved final edits of the manuscript. All authors approved the final version of the paper to be published.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.