Article Text

Abstract
Introduction PROOF (a Prospective Observational Registry to Describe the Disease Course and Outcomes of Idiopathic Pulmonary Fibrosis) is an ongoing, observational registry initiated in 2013 with the aim of collecting real-world data from patients with idiopathic pulmonary fibrosis (IPF). Here, we present comprehensive baseline data, which were collected from patients on registry inclusion.
Methods Patients with IPF were enrolled across eight centres in Belgium and Luxembourg. Baseline data collected included demographics, diagnostic information and clinical characteristics, including lung function and health-related quality of life. Data on comorbidities and prescribed medication were also collected.
Results A total of 277 patients were enrolled in the PROOF registry. At inclusion, 92.8% and 6.5% of patients had a definite or probable diagnosis of IPF, respectively. Mean per cent predicted forced vital capacity and carbon monoxide diffusing capacity were 80.6% and 46.9%, respectively. Mean St. George’s Respiratory Questionnaire total score was 47.0, and mean Cough-Visual Analogue Scale score was 30.5 mm. The most prevalent comorbidities reported at inclusion were gastrointestinal disorders (50.2%), including gastro-oesophageal reflux disease (47.3%) and metabolism and nutrition disorders (39.7%). At inclusion, 67.2% and 2.2% of patients were prescribed pirfenidone and nintedanib, respectively, with treatment initiated either prior to, or at the time of, inclusion. Medication prescribed concomitantly with pirfenidone included antihypertensives (54.8%), statins (37.1%) and prophylactic antithrombotics/anticoagulants (36.6%).
Conclusion The PROOF registry provides valuable demographic and clinical data from a real-world population of patients with IPF in Belgium and Luxembourg, demonstrating the high burden of comorbidities and prescribed medication in these patients. Longitudinal data from this patient population will be investigated in future analyses.
Trial registration PROOF is registered with the relevant authorities in Belgium and Luxembourg, with registration to Comité National d’Éthique et de Recherché (CNER) N201309/03 – 12 September 2013 and a notification to Comité National de Protection des Données (CNDP).
- interstitial fibrosis
- rare lung diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0
Statistics from Altmetric.com
Key messages
What information can the observational PROOF registry provide on clinical characteristics, comorbidities and medication use in a real-world population of patients with idiopathic pulmonary fibrosis (IPF)?
The PROOF registry provides valuable real-world data from a population of patients with IPF in Belgium and Luxembourg, demonstrating relatively preserved lung function and quality of life, despite a high burden of comorbidity and prescribed medication.
In clinical trials enrolling patients with IPF, inclusion/exclusion criteria relating to lung function and medical histories are very restrictive, and do not always provide an accurate reflection of patient populations in daily clinical practice.
The PROOF registry provides an important opportunity to characterise a real-world population of patients with IPF with a range of clinical characteristics, comorbidities and prescribed medications.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a debilitating, irreversible, fatal, fibrosing lung disease.1 2 Progressive symptoms including cough and shortness of breath lead to increasing limitations on daily activities and a growing dependence on family or caregivers.3–6
Two antifibrotics are currently available for the treatment of patients with IPF: pirfenidone and nintedanib, which were approved in Europe in 2011 and 2015, respectively.7 8 In a pooled analysis of three phase III clinical trials, ASCEND (study 016: Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis; NCT01366209) and CAPACITY (studies 004 and 006: Randomized, Double-Blind, Placebo Controlled, Phase 3 Studies of the Safety and Efficacy of Pirfenidone in Patients With Idiopathic Pulmonary Fibrosis; NCT00287716 and NCT00287729), pirfenidone significantly reduced lung function decline versus placebo over 52 weeks.9 Similarly, in the INPULSIS trials of nintedanib (INPULSIS 1 and 2: 52 Weeks, Double Blind, Randomized, Placebo-controlled Trials Evaluating the Effect of Oral BIBF 1120, 150 mg Twice Daily, on Annual Forced Vital Capacity Decline, in Patients With Idiopathic Pulmonary Fibrosis; NCT01335464 and NCT01335477), nintedanib significantly reduced lung function decline versus placebo over 52 weeks.10
Although these studies provide valuable information regarding the efficacy and safety of antifibrotic treatments in patients with IPF, it should be acknowledged that the inclusion and exclusion criteria were restrictive. In ASCEND, CAPACITY and INPULSIS, patients with per cent predicted forced vital capacity (FVC) <50% or per cent predicted carbon monoxide diffusing capacity (DLco) <30% (ASCEND and INPULSIS) or <35% (CAPACITY) were excluded.10–12 Furthermore, patients with unstable or deteriorating cardiac or pulmonary disease, or those who were receiving certain prescribed medications, were also excluded from these trials. However, real-world evidence suggests that comorbid conditions are prevalent in patients with IPF in clinical practice, with results from the European MultiPartner IPF Registry (EMPIRE) reporting that 86% of patients with IPF had a comorbid diagnosis.13 These data suggest that there is a need to further investigate characteristics and treatment outcomes in real-world populations of patients with IPF, which may be more representative of clinical practice compared with clinical trial populations.
Longitudinal patient registries are recognised as playing an important role in providing long-term data on the clinical course of IPF and the impact of treatment in the real-world clinical setting.14–16 The PROOF registry was initiated in 2013 to monitor disease progression in a real-world European population of patients with IPF17 and is currently one of the most comprehensive sources of data on patients with IPF treated with pirfenidone in the real-world setting.
Here, we present baseline data collected from patients on enrolment into the PROOF registry, including patient demographics, clinical characteristics such as lung function and health-related quality of life (HRQoL), as well as comorbidities and prescribed medications.
Methods
Registry design
The PROOF registry is an ongoing observational study initiated in October 2013 to monitor disease progression in a real-world population of patients with IPF.
Patients were enrolled across seven centres in Belgium and one centre in Luxembourg between October 2013 and January 2016. The majority of diagnoses took place in one of the ‘expert centres’, which had an experienced multidisciplinary team (MDT) present; however, subsequent follow-up after diagnosis was performed jointly in the expert centres and the other centres. Eligible patients were over 18 years of age, with a MDT diagnosis of definite or probable IPF according to 2011 American Thoracic Society/European Respiratory Society guidelines.2 Patients with a history of environmental, occupational or drug exposures were eligible for inclusion in the PROOF registry if MDT discussions excluded hypersensitivity pneumonitis or other interstitial lung diseases (ILD) with known cause. Patients were excluded if they were enrolled in a clinical trial at the time of inclusion in the PROOF registry or if they were unable to give informed consent.
The PROOF registry was conducted in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Guidelines for Good Clinical Practice and local legal and regulatory requirements. Patients were required to provide informed consent prior to inclusion.
Baseline demographics and clinical characteristics
Baseline data were collected from patients on inclusion in the PROOF registry. Information on IPF diagnosis was also collected at the baseline visit, including any diagnostic tests performed prior to registry inclusion to reach the final diagnosis (definite or probable IPF).
Baseline patient demographics including age, gender and smoking status were recorded. Lung function was assessed using per cent predicted FVC and per cent predicted DLco. Other clinical assessments included 6 min walk distance (6MWD) and the Gender, Age and Physiology (GAP) Index.18
Self-reported HRQoL, generic health status and cough severity were assessed using the St. George’s Respiratory Questionnaire (SGRQ), the EuroQoL-Visual Analogue Scale (EQ-VAS), the EuroQoL-5 Dimensions Health Questionnaire (EQ-5D) and the Cough-Visual Analogue Scale (Cough-VAS).19–21 The SGRQ has been confirmed as an appropriate tool to measure HRQoL in patients with IPF across three domains: symptoms, activity and impacts. Scores are weighted to provide a total score from 0 to 100, with higher scores indicating greater impairment.19 22 23 The EQ-VAS was used by patients to rate their overall health status on a linear visual scale, with 0 mm representing ‘worst health imaginable’ and 100 mm representing ‘best health imaginable’.20 The EQ-5D measures impairment across five dimensions related to generic health status: mobility, self-care, usual activities, pain/discomfort and anxiety/depression, with patients rating their impairment on a categorical scale (no problems, slight problems, moderate problems, severe problems, extreme problems).20 The Cough-VAS is a linear visual scale on which patients are asked to indicate the severity of their cough, with 0 mm representing ‘no cough’ and 100 mm representing ‘worst cough ever’.21
Current medications prescribed for the treatment of IPF were recorded at baseline, including those initiated prior to inclusion in the registry and those initiated at the time of inclusion into the registry. Comorbid medical conditions were also recorded at baseline, as were any medications prescribed for indications other than IPF, using a combination of medical records and self-reporting from patients. Data were collected using electronic case report forms (eCRFs), which were completed by both patients and healthcare practitioners at baseline and every 3–6 months thereafter, per routine clinical practice.
Data analysis
The baseline data reported in this manuscript were reviewed at a cut-off date of March 2017.
A programmed database received all information collected in the eCRFs, and automated edit checks were conducted. A contract research organisation was responsible for the management of data, including data quality checks, and was also required to produce a Data Review Strategy to highlight the quality check method performed on the data.
Baseline data were summarised using descriptive statistics. Comorbid medical conditions were coded according to the Medical Dictionary for Regulatory Activities and summarised descriptively.
Results
Patient demographics and diagnostic information
A total of 277 patients were enrolled in the PROOF registry and provided baseline data. The majority of patients were men (76.9%), white (92.1%) and ever-smokers (66.8% former smokers, with only 6.5% current smokers (table 1)).
Patient demographics at registry inclusion.
Mean (SD) age was 69.6 (8.6) years. A family history of pulmonary fibrosis was reported in 7.6% of patients. A total of 38.6% of patients reported any employment exposure, most commonly to asbestos (11.6%), paint and/or chemicals (9.4%) or metal dust (8.7%). The most common signs and symptoms of IPF present at baseline were Velcro rales (78.0%), breathlessness (67.9%), cough (66.8%) and clubbing (19.1%).
A total of 92.8% of patients enrolled in the PROOF registry had a ‘definite’ diagnosis of IPF, and a diagnosis of ‘probable’ IPF was recorded in 6.5% of patients (table 2). Data on the presence or absence of MDT diagnosis were available for 276 patients, of whom 270 (97.8%) received an MDT diagnosis. High-resolution CT (HRCT) images were obtained at diagnosis in 97.5% of patients, and bronchoalveolar lavage was performed in 80.9% of patients. Open surgical lung biopsy, video-assisted thoracoscopic surgery (VATS) and cryobiopsy were performed in 13.7%, 14.8% and 1.8% of patients, respectively. Transbronchial biopsy was not reported to have been performed in patients included in the PROOF registry.
Diagnostic data and clinical characteristics collected at registry inclusion.
The median time between patients first becoming aware of their symptoms and a diagnosis of IPF was 281 days, with 14 patients receiving a diagnosis of IPF prior to reporting any symptoms.
Clinical characteristics
At baseline, mean (SD) per cent predicted FVC was 80.6% (19.9), mean (SD) per cent predicted DLco was 46.9% (13.8) and mean (SD) 6MWD was 426.2 m (130.8; table 2). The majority of patients enrolled in PROOF were GAP stage I (38.7%) or II (40.8%).
Mean (SD) SGRQ total score at baseline was 47.0 (20.2), with the greatest impairment (mean subscore [SD]) reported on the activity subscore (62.2 [25.3]), followed by the symptoms (48.2 [22.4]) and impacts (36.4 [21.8]) subscores (table 3). When asked to describe their current health, the majority of patients reported ‘fair’ (52.9%) or ‘good’ (29.7%) health, while ‘poor’ or ‘very poor’ health were reported by 12.0% and 3.9% of patients, respectively.
Health-related quality of life at registry inclusion.
Mean (SD) EQ-VAS score at baseline was 61.1 mm (19.2). Across the EQ-5D components, patients reported greatest impairment on the mobility dimension, with moderate, severe or extreme problems reported by 51.7% of patients (figure 1). No problems or slight problems were reported by the majority of patients across the remaining EQ-5D dimensions of self-care (80.0%), anxiety/depression (74.5%), pain/discomfort (65.2%) and usual activities (56.8%) (figure 1). Cough severity, measured using mean (SD) score on the Cough-VAS, was 30.5 mm (25.2) (table 3).

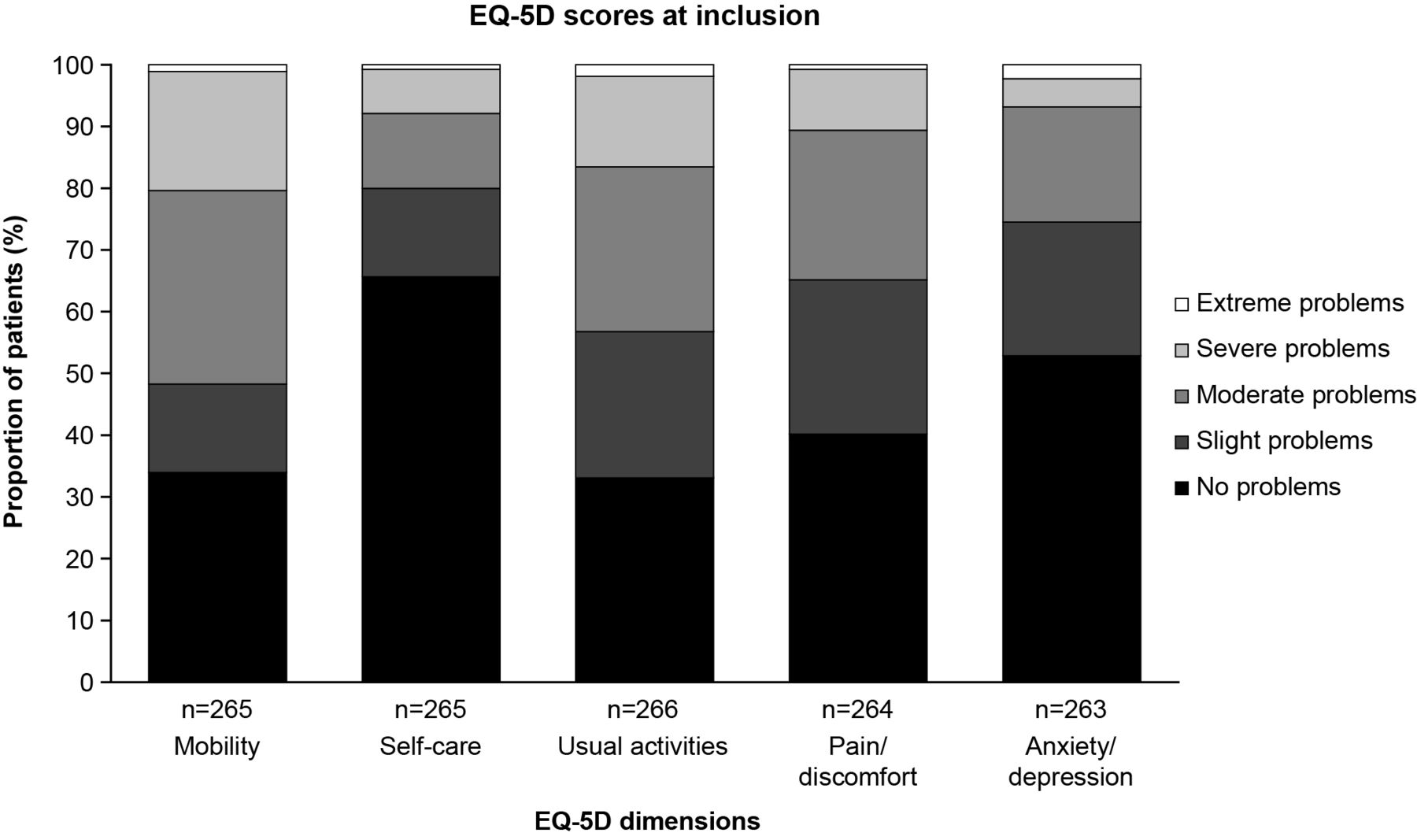{kind=link}
Proportion of patients reporting problems across each dimension of the EQ-5D at registry inclusion. EQ-5D, EuroQol-5 Dimensions Health Questionnaire.
Comorbidities and prescribed medications
Gastrointestinal disorders and metabolism and nutrition disorders were the most prevalent comorbidities reported at baseline, present in 50.2% and 39.7% of patients, respectively, including 47.3% of patients who reported gastro-oesophageal reflux disease (GERD). Musculoskeletal disorders were present in 18.8% of patients, while respiratory, thoracic and mediastinal disorders were present in 19.9% of patients, including 4.0% of patients who reported pulmonary hypertension and 4.0% of patients who reported emphysema. Other specific common comorbidities included vascular disorders (37.9%), hypertension (33.2%) and cardiac disorders (27.8%; table 4).
Comorbidities recorded at registry inclusion.
Overall, 69.3% of patients were prescribed an antifibrotic at baseline, initiated either prior to, or at the time of, inclusion in the registry, with 186 and 6 patients prescribed pirfenidone and nintedanib, respectively (table 5). Among those patients prescribed pirfenidone, the most frequently co-prescribed medications were antihypertensives, which were prescribed in 54.8% of patients receiving pirfenidone. This was followed by statins (37.1%) and prophylactic antithrombotics or anticoagulants (36.6%), for example, aspirin or low-dose low-molecular-weight heparins (table 5). We also investigated the concomitant prescribing of pirfenidone and antidepressants, neuroleptics or hypnotics based on the potential interaction between pirfenidone and some of these medications. Concomitant antidepressants, neuroleptics or hypnotics were prescribed in 2.7% of patients receiving pirfenidone (duloxetine, n=1; prothipendyl, n=1; amitriptyline, n=3; escitalopram, n=1; lorazepam, n=1; trazodone, n=1; some patients received more than one of the medications listed).
Prescribed medications recorded at registry inclusion.
Discussion
The PROOF registry collects data from a real-world cohort of patients with IPF in Europe. Unlike clinical trials, which have strict inclusion/exclusion criteria relating to disease severity, comorbidities and prescribed medications, the PROOF registry provides an opportunity to characterise a real-world population of patients. Compared with clinical trials, registry data are more likely to reflect the range of disease severities and complex medical histories that clinicians observe in clinical practice. In the present manuscript, we report a comprehensive summary of baseline data from patients enrolled in the PROOF registry, including patient demographics and clinical data such as lung function and HRQoL. In addition, we present data demonstrating the high burden of comorbidities and prescribed medication in this population.
Further to providing comprehensive clinical data, the PROOF registry also provides an opportunity to collect information on the diagnosis of IPF in a real-world population. In the PROOF registry, the median length of time between the appearance of the first symptoms of IPF and subsequent diagnosis was 281 days. Delays in the diagnosis of IPF have been reported elsewhere, with a qualitative European survey of patients with IPF reporting that 87% of patients waited more than 1 year to receive a diagnosis.5 In Belgium and Luxembourg, the patient journey to diagnosis can be relatively rapid once patients are referred to specialist centres. However, it is well known that the lack of awareness regarding IPF among primary care physicians and difficulty accessing specialist centres can lead to delays in referral and diagnosis,5 24 although this cannot be evaluated using registry data such as those from PROOF. Misdiagnosis with other respiratory disorders has also been identified as a key reason for delayed diagnosis of IPF.5 Indeed, in the PROOF registry, the non-specific symptoms of cough and breathlessness were present at baseline in 66.8% and 67.9% of patients, respectively. However, Velcro rales, which are a common but non-specific early feature of IPF,25 were the most common finding present at the baseline visit of PROOF, recorded in 78% of patients. Although we could not confirm if this feature of IPF was present at the beginning of the diagnostic process for patients included in the PROOF registry, it is possible that targeting education on Velcro rales to primary care physicians could help to reduce delays in referral and diagnosis.
Once patients with suspected ILD are referred to a specialist centre, international treatment guidelines recommend that a diagnosis of IPF be made based on the presence of a usual interstitial pattern (UIP) on HRCT images of the chest.2 In patients without clear UIP, a biopsy may be required to confirm a diagnosis of IPF.2 Almost a third of patients in the PROOF registry underwent lung biopsy (open surgical, VATS or cryobiopsy) as part of the IPF diagnostic process, which is a greater proportion compared with observations from other IPF registries. For example, in the Finnish IPF registry, the Australian IPF registry and the Swedish IPF registry, the rate of lung biopsy was approximately 20%.26–28 The high biopsy rate observed in the PROOF registry may be partially explained by evidence suggesting that patients with less advanced IPF are less likely to demonstrate definite UIP on HRCT at the time of diagnosis.29 The potential relationship between less advanced disease and the likelihood of definite UIP on HRCT is relevant because many patients in the PROOF registry had relatively preserved lung function, with 39% of patients in GAP stage I and mean per cent predicted FVC of 80.6% at baseline. Another factor to be considered, which may partially explain the high biopsy rate observed in the PROOF registry, is the time period at which patients were enrolled compared with other IPF registries. Practices used in biopsy and HRCT during the diagnosis of IPF are ever evolving, for example, the Fleischner Society White Paper outlines the process of forming working diagnoses in patients with a probable UIP pattern and a consistent clinical context. In these cases, a surgical lung biopsy is not required.30 However, the 2018 international diagnostic guidelines provide a conditional recommendation for surgical lung biopsy in patients with probable UIP, a recommendation that has provoked much discussion.31 32 These changes in diagnostic practice over time and variations in interpretation are likely to affect comparisons between real-world populations, particularly in countries including Belgium where diagnosis of IPF with a high CI is necessary (and was necessary at the time of enrolment in the PROOF registry) for reimbursement of antifibrotics. Going forward, uptake and application of the new recommendations may not be immediate or uniform and may be one factor behind inter-registry variations in the proportions of patients undergoing biopsies.
The observation that patients in the PROOF registry had relatively preserved lung function is similar compared with other IPF registries across the world, which have reported mean FVC values between 68% and 81%.26–28 33–35 Similarly, although patients enrolled in PROOF reported impaired HRQoL, as would be expected in a population of patients with IPF, the majority of patients described their health as ‘fair’ or ‘good’ based on the results of the SGRQ. Data from the German INSIGHTS-IPF (INvestigating SIGnificant Health TrendS in idiopathic pulmonary fibrosis) registry have previously demonstrated that FVC and GAP Index are significantly associated with HRQoL in patients with IPF.36 Similarly, in the PROOF registry, lung function and HRQoL were both relatively preserved.
Another significant predictor of impaired HRQoL in patients with IPF is the presence of comorbidities, with an increasing number of comorbidities associated with greater impairment reported using the SGRQ total score.36 In the PROOF registry, a wide range of comorbidities were reported at baseline. The most prevalent comorbidities were gastrointestinal disorders, in particular GERD, which was present in nearly half of all patients enrolled in PROOF. Other prevalent conditions included metabolism and nutrition disorders and vascular disorders. These results are similar to those from other IPF registries, with GERD and vascular disorders shown to be prevalent among patients enrolled in both the Australian IPF registry and the INSIGHTS-IPF registry.28 33 Interestingly, the prevalence of pulmonary hypertension appeared to be lower in the PROOF registry compared with other real-world registries. For example, in both the Australian IPF registry and the INSIGHTS-IPF registry, pulmonary hypertension at baseline was suspected or confirmed in nearly 20% of patients compared with only 4% in PROOF.28 33 It is possible that the observed differences in the prevalence of pulmonary hypertension might be partially explained by a combination of patients having less advanced disease in the PROOF registry compared with other registries, the use of different data collection modalities, for example, self-reported patient questionnaires used to collect comorbidity data in the Australian IPF registry, or differences in diagnostic technique, for example, differences in the routine performance of echocardiography and availability of right heart catheterisation and underdetection due to differences in the sensitivity of the diagnostic modality used.
As would be expected in a population of patients with a high burden of comorbidity, the majority of patients in the PROOF registry were prescribed one or more medications at baseline. The large proportion of patients in PROOF who were prescribed antithrombotics/anticoagulants or statins is of particular interest. Based on previous research investigating clinical outcomes in patients with IPF enrolled in the placebo arms of ASCEND or CAPACITY who were treated with anticoagulants or statins for non-IPF indications, these post-hoc analyses have suggested that while statins may have some beneficial effects on disease-related outcomes in patients with IPF,37 anticoagulants might be associated with an increased risk of IPF-related mortality over 52 weeks.38 In the absence of clinical trials investigating the use of multiple medications in patients with IPF, registries such as PROOF will provide an invaluable opportunity to study the relationship between prescribed medications and longitudinal outcomes in patients with IPF.
In the PROOF registry, a total of 69.3% of patients were treated with an antifibrotic at baseline, initiated prior to, or at the time of, registry inclusion. This is a higher proportion than has been previously reported in other registries, with 44.2% and 23.0% of patients receiving antifibrotic treatment at inclusion in the INSIGHTS-IPF registry and Australian IPF registry, respectively.28 33 Licensing and reimbursement criteria are key factors in influencing antifibrotic prescribing patterns across different regions. For example, in the PROOF registry, the higher proportion of patients prescribed pirfenidone versus nintedanib was largely driven by the fact that while pirfenidone was approved for the treatment of patients with IPF in Europe in 2011, nintedanib was not approved until 2015.7 8 In addition, enrolment into the PROOF registry began in 2013, at a time when pirfenidone treatment was reimbursed in patients with ‘mild to moderate’ IPF in Belgium and Luxembourg.
Although the most recent international consensus guidelines for the treatment of IPF recommend that the majority of patients with IPF should be considered for treatment with an antifibrotic,39 it is possible that many clinicians might be reluctant to prescribe antifibrotics in patients receiving multiple prescribed medications40 and with multiple comorbid conditions. In the absence of clinical trials, the PROOF registry provides a valuable opportunity to investigate the use of antifibrotics in a real-world population with a high level of comorbidity and prescribed medication. Of particular interest, we observed that many patients enrolled in the PROOF registry who were prescribed pirfenidone at the baseline visit were also prescribed other medications concomitantly. The most commonly co-prescribed medications in patients receiving pirfenidone were antihypertensives, statins and prophylactic antithrombotics/anticoagulants.
The PROOF registry provides valuable data from a real-world population of patients with IPF. However, the potential limitations of the PROOF registry should also be considered. For example, the PROOF registry enrolled a relatively small number of patients (N=277) across a limited geographical area. The global prescribing pattern of antifibrotics varies by geographical area depending on factors such as reimbursement or prescribing guidelines; for example, in Belgium, antifibrotic prescribing is limited to centres with a vast amount of experience in the clinical diagnosis and management of IPF. Therefore, it is possible that results from the PROOF registry may not be representative of populations of patients with IPF in different countries, thus limiting cross-registry comparisons. In addition, it may be the case that patients with comorbid life-threatening conditions, for example, lung cancer or advanced heart failure, or patients with more advanced disease, are not referred to centres of excellence for the treatment of IPF due to concerns regarding treatment tolerability or lack of reimbursement in patients with more advanced disease. These patients would not have been included in the PROOF registry and this could affect the comparison of results from PROOF with the real-world patient population. The diagnoses of the majority of patients included in the PROOF registry at one of three ‘expert centres’ could also be considered a limitation, as these centres had an experienced MDT present which may not be representative of the real world. In addition, we are restricted in evaluating how comparable the diagnoses of IPF in the PROOF registry are versus other real-world populations as we do not have further data regarding the breakdown of HRCT and lung biopsy patterns and subsequent diagnoses. A further limitation is that some patients participating in the PROOF registry were diagnosed with IPF prior to enrolment in the registry; therefore, some of the diagnostic data captured in the PROOF registry may have been retrospective. It should also be acknowledged that the PROOF registry is not designed as a clinical trial; therefore, patients were not randomised to receive treatments as per usual clinical practice, and so different treatments may not be comparable with respect to outcomes.
In conclusion, the PROOF registry provides valuable information on the characteristics of patients with IPF in a real-world setting. Baseline data collected from patients enrolled in the PROOF registry demonstrated modest delays in diagnosis, which were shorter than have been previously reported by patients within Europe, with relatively preserved lung function and HRQoL. A high burden of comorbidities and prescribed medications was evident in PROOF, with many patients receiving concomitant medications in addition to pirfenidone, most commonly antihypertensives and anticoagulants/antithrombotics. Whereas clinical trials have focused on patients with limited comorbidity, the PROOF registry will be able to provide data more representative of patients in clinical practice. Future analyses in this real-world cohort will investigate longitudinal outcomes in patients with IPF with a range of comorbidities and prescribed medications.
Acknowledgments
The authors would like to thank Gheorghita Amand (research assistant), Els De Dycker (data manager), Gerda Verbessem (data manager), Yuan-Chi (Daisy) Lee and Yifeng (Jenny) Chia (statistical and analytic support) for all of their assistance. Medical writing support was provided by Marianne Wells and Ceilidh McConnachie of CMC AFFINITY, a division of Complete Medical Communications Ltd, Glasgow, UK, funded by F. Hoffmann-La Roche, Ltd.
References
Footnotes
Contributors WAW, BB, MS, HS, CD: were involved in the conception and design of the PROOF registry. WAW, BB, MS, HS, CD: conducted data collection. KB: conducted data analysis. All authors contributed towards the interpretation of the results, and actively participated in drafting and revising this manuscript. All authors were involved in the final approval of the manuscript version to be published and agree to be accountable for all aspects of the work.
Funding This study was sponsored by Genentech, Inc. and F. Hoffmann-La Roche, Ltd.
Competing interests WAW, BB, CD and HS have received consulting and/or lecture fees from F. Hoffman-La Roche, Ltd. and Boehringer Ingelheim. KUK and KB are employees of Roche-Genentech and hold shares.
Patient consent Not required; this article does not contain personal medical information about an identifiable individual.
Ethics approval Comité National d’Éthique et de Recherché (CNER) N201309/03 Comité National de Protection des Données (CNDP).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Qualified researchers may request access to individual patient level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available here (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).