Article Text

Abstract
Introduction Pneumothorax is a common clinical problem. Primary spontaneous pneumothorax (PSP) occurs in otherwise fit young patients, but optimal management is not clearly defined and often results in a long hospital stay. Ambulatory treatment options are available, but the existing data on their efficacy are poor. The Randomised Ambulatory Management of Primary Pneumothorax trial is a multicentre, randomised controlled trial comparing ambulatory management with standard care, specifically designed to safely and effectively reduce hospital stay.
Methods and analysis 236 patients with PSP will be recruited from UK hospitals. Patients will be randomised 1:1 to treatment to either the ‘Intervention’ arm (ambulatory device; Rocket Pleural Vent) or the ‘Control’ arm (aspiration ± standard chest drain insertion). Patients will be followed up for a total of 12 months to assess recurrence rates. The primary outcome is total length of stay in hospital (including readmissions) up to 30 days postrandomisation. The secondary outcomes are pain and breathlessness scores, air leak measurement and radiological evidence (on CT scanning) of emphysema-like changes, compared with short-term and long-term outcomes, respectively, and health economic analysis.
Ethics and dissemination The trial has received ethical approval from the National Research Ethics Service Committee South-Central Oxford A (15/SC/0240).
Trial registration number ISRCTN79151659
- pleural disease
- pneumothorax
- ambulatory
- outpatient
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Pneumothorax—air in the pleural space—is a common pathology. Primary spontaneous pneumothorax (PSP) conventionally refers to patients developing a pneumothorax, in the absence of trauma, with no underlying established lung pathology. PSP occurs in ~3000 patients per year in the UK.1–3 A minority can be managed conservatively with close observation only. A currently recruiting trial is assessing conservative versus interventional management.4 However, many patients will require an intervention to re-expand the lung. In most patients ‘aspiration’ of the trapped air using a cannula and syringe is considered, but more than 50% will require treatment with insertion of a chest drain and a standard underwater seal. The average duration of inpatient stay of patients admitted for drainage is 6–8 days.5
Ambulatory management of patients with PSP could potentially remove the need for long hospital admission and outpatient treatment. Reducing the need for chest drains with bulky underwater systems may allow patients to be more mobile and facilitate earlier discharge. A ‘Heimlich valve’ (one-way valve connected to a chest drain, rather than a bulky underwater seal) has been previously proposed, in the form of either one-way valves attached to standard chest drains, as well as the relatively new ‘pocket’ devices in which the drainage catheter and one-way valve are integrated into a single device. A systematic review of 18 studies using ambulatory management reported an overall success rate of 85.8% and a successful outpatient management in 77.9% with ‘few complications’. However, the evidence was of poor quality with a high risk of bias, with only two small randomised trials and with the remainder being retrospective case series.6
The Randomised Ambulatory Management of Primary Pneumothorax (RAMPP) trial aims to provide randomised controlled trial data to definitively answer the question of whether an ambulatory management of patients with PSP can safely and effectively reduce hospital stay. The ambulatory device used in this trial is Rocket Pleural Vent (Rocket Medical, UK), specifically designed for treatment of pneumothorax.
At present, there exist no good predictive data to guide management of either short-term failure of initial management (with ongoing air leak or non-re-expanding lung) or long-term with recurrence of pneumothorax. Therefore, the RAMPP trial will also prospectively collect clinical data, including daily air leak measurement and Computed Tomography (CT) imaging on all patients to potentially generate a predictive tool allowing the selective and targeted treatment of patients according to likely outcome, resulting in more personalised treatment (and intended to be published separately from the main findings of the study).
This study, evaluating the randomised ambulatory management of primary pneumothorax (RAMPP trial), is a multicentre, open-label, randomised controlled trial comparing ambulatory management with standard care (aspiration ± standard chest drain insertion) with an observational cohort study of patients not requiring an intervention. The trial is sponsored by the University of Oxford and is being managed by the Oxford Respiratory Trials Unit (ORTU). Data management is undertaken by ORTU.
The trial is registered on the International Standardised Randomised Controlled Trial Registry. The study is included in the National Institute for Health Research Clinical Research Network portfolio for both respiratory and emergency medicine.
The following is the primary research question: for patients with PSP, can use of an ambulatory device and treatment strategy significantly reduce hospital stay within the first 30 days of initial presentation?
The following are the secondary research questions:
Is ambulatory care and early discharge safe and cost-effective?
Is patient experience improved with an ambulatory device, including pain of procedure, breathlessness, quality of life assessments (EuroQol EQ-5D-5L) and time to return to working status?
What is the recurrence rate of pneumothorax over 6 and 12 months?
Does radiological evidence (on CT scanning) of emphysema-like changes and inflammation predict long-term outcome (ie, recurrence of pneumothorax within 12 months)?
Can digitally measured air leak and its evolution over treatment predict short-term clinical trajectory in patients with pneumothorax, including failure of treatment (ie, prolonged air leak or non-expanded lung)?
Study design
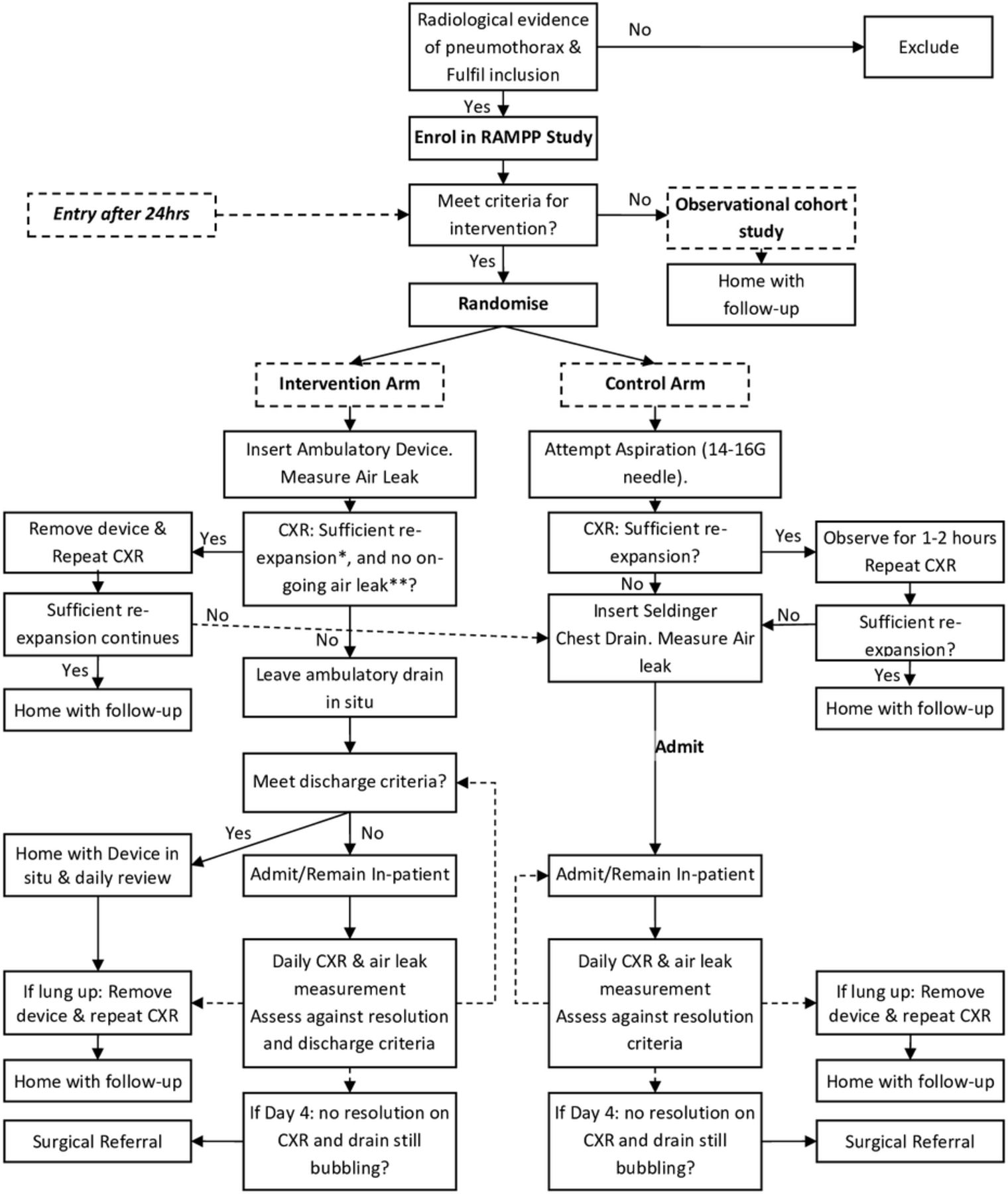
Two hundred and thirty-six patients with PSP requiring an intervention will be recruited from UK centres (see online supplementary appendix A for the list of UK centres). Patients will be randomised 1:1 to either insertion of an ambulatory device (Rocket Pleural Vent) or to standard care (aspiration ± standard chest drain insertion) as per the British Thoracic Society (BTS) guidelines.7 Patients who do not require an intervention will be invited to participate in an observational cohort study, with the results of this cohort published separately.
Supplemental material
Patient involvement
Patients were involved in the design and conduct of this research. During the feasibility stage, priority of the research question, choice of outcome measures and methods of recruitment were informed by discussions with patients while in hospital and at follow-up in clinic. A patient representative is an independent member of the trial steering committee.
Participant selection
All patients presenting with spontaneous pneumothorax to emergency departments or acute medical teams will be assessed for eligibility. Patients may only be randomised once into the RAMPP trial. However, if a patient initially enrolled in the observational cohort subsequently requires treatment, they can be re-enrolled (and reconsented) for randomisation into the interventional component of the trial.
Inclusion criteria
Presenting with spontaneous pneumothorax as confirmed by a chest radiograph (CXR) or a CT scan.
Age between 16 and 55 years old.
Ability to consent to participation.
Exclusion criteria
Known or suspected underlying lung disease (including >20 pack-year smoking history).
Evidence of tension pneumothorax (defined as clinical or radiographic evidence of significantly increased intrapleural pressure causing haemodynamic compromise requiring urgent decompression). These patients should be treated immediately as medical emergencies.
Women who are pregnant or lactating.
Inability to consent or comply with the trial requirements.
Contraindication to thoracic procedure (only applies to patients being enrolled into the intervention or control arms, ie, not observational cohort).
Any other significant disease or disorder which, in the opinion of the investigator, may either put the participants at risk because of participation in the trial, or may influence the result of the trial, or the participant’s ability to participate in the trial.
‘Mild’ asthma is not considered an exclusion criterion. Patients with a diagnosis of asthma in childhood/young adulthood who do not require the use of a regular ‘preventer’ inhaler (ie, inhaler containing a steroid or long-acting beta-agonist) and have never been hospitalised due to asthma remain eligible for participation in this study.
Patients requiring intervention can be enrolled and randomised up to 24 hours after presentation, as long as they are still an inpatient, have an ongoing symptomatic pneumothorax despite initial intervention (eg, patients initially treated with aspiration overnight) and now require chest drain insertion. Patients not requiring intervention can be enrolled into the observational cohort up to 2 weeks after their initial presentation.
Informed consent
Once an eligible patient is identified and agrees to participate in the study, informed written consent will be obtained by the principal investigator or other suitably qualified delegated personnel. As pneumothorax is an acute medical problem, it would not be appropriate to wait the usual 24 hours to allow patients time to read the patient information leaflets, prior to intervention. However, ideally the patient should still be given a reasonable short period of time to read, digest and ask questions about the study, prior to an approach for consent.
Randomisation and blinding
Patients will be randomised using a centralised, web-based randomisation system with each patient being assigned a unique trial number. Minimisation with a residual random component will occur, for the minimisation factors of (1) recruiting centre and (2) size of pneumothorax (≥4 cm vs <4 cm) at presentation.
Due to the nature of the interventions, patients and clinicians cannot be blinded to allocation, and therefore code-breaking is not required for this trial. However, the objective ‘fitness for discharge’ data will be blind-reviewed after the trial by an independent assessor blind to treatment arm (ie, objective blind outcome assessment) and compared with actual time spent in hospital at study end. Recurrence rates at 12 months will be blindly assessed by an independent assessor by reviewing patient CXRs and hospital records for readmissions at study end. In addition, the clinician responsible for making a decision to discharge a patient will be blinded to the air leak measurements recorded as part of the trial protocol.
Interventions
Once the patient has been identified as having a PSP by CXR, the decision to intervene will be made on the basis of the current BTS guidelines: large (interpleural distance at the level of hilum ≥2 cm) and/or symptomatic patients will be randomised to the following:
Intervention arm: ambulatory device (Rocket Pleural Vent)
An ambulatory device (Pleural Vent, Rocket Medical) should be inserted immediately after randomisation, using local anaesthetic. Researchers and local clinicians will be trained in inserting the device. The patient is then observed for 1–2 hours to check for clinical stability, after which a repeat CXR is performed (see figure 1).

{kind=link}
Study flow chart. CXR, chest radiograph; RAMPP, Randomised Ambulatory Management of Primary Pneumothorax.
If CXR shows insufficient re-expansion of the lung†, the pleural vent should remain in place and the patient can be discharged with device in situ if the patient fulfils the ‘Fitness for discharge’ criteria, which are all of the following:
Patient agreement.
Clinically stable cardiorespiratory observations (oxygen saturation, respiratory and heart rate, blood pressure).
No increase in size of pneumothorax (since last review).
Not requiring oxygen or other ventilator assistance.
Patient is mobile and independent to self-care.
Written information on point of contact if there are concerns and follow-up plan.
Patient lives with a responsible person at home and is able to help the patient if required.
Any patient not meeting these criteria should remain as an inpatient and be reviewed daily.
If discharged, patients should be seen every 1–2 days until day 4; ideally daily, but once over the weekend is sufficient if clinically stable. At each review, if there is sufficient re-expansion of the lung† and no ongoing air leak clinically††, the responsible clinician can consider removing the device and the patient can be discharged home. As standard practice, a postremoval CXR should be performed to ensure that the lung has not recollapsed. If the CXR shows that the pneumothorax has recurred, the patient should be admitted to the respiratory ward with consideration of placement of a standard chest drain.
*‘Sufficient re-expansion’ is defined as complete or almost complete re-expansion (only a very small [<1 cm] rim of air apically) on CXR.
**Ongoing air leak assessed by attempted aspiration through the device using a syringe and connector: if the device is patent (as assessed by movement of the inbuilt diaphragm) but unable to aspirate (ie, draw air back through syringe), then there is no ongoing air leak; if able to aspirate air freely, then there is ongoing leak and active pneumothorax.
Control arm: standard management as per the BTS guidelines
Treatment in this arm is as per the BTS Pleural Guidelines 2010.7 In brief, needle aspiration (NA) should be attempted under local anaesthetic using 14–16 gauge cannula and syringe, aspirating a maximum of 2.5 L. The patient should be observed for 1–2 hours to check for clinical stability and a repeat CXR should be performed. If the repeat CXR after shows sufficient re-expansion of the lung, the patient can be discharged home. However, if the lung has not sufficiently re-expanded, then a small-bore chest drain (≤14F) should be inserted and attached to an underwater seal, and the patient admitted to hospital.
Although initial aspiration is recommended, the responsible clinician may decide to proceed directly to chest drain insertion and admission at their discretion (and according to the BTS guidelines).
Decisions regarding drain removal are as per the standard practice at the participating centre, but typically will include the following: no further air leak as demonstrated by a non-bubbling chest drain and full lung expansion on CXR. As standard practice, a postremoval CXR should be performed to ensure that the lung has not collapsed. ‘Fitness for Discharge’ criteria for discharge will be conducted daily to provide equality between treatment arms (criteria as above).
Study assessments
This study will use a web-based, dedicated and validated clinical trial database designed for remote electronic data capture (OpenClinica). All baseline demographic data, daily clinical observations, adverse events (AEs) and healthcare usage data will be uploaded directly onto OpenClinica. The full schedule of study procedures is found in online supplementary appendix B.
All patients will document a visual analogue scale (VAS) score for thoracic pain and breathlessness at baseline (day 0, prior to procedure and postprocedure) and daily for the next 4 days of assessment. Quality of life questionnaires (EQ-5D-5L) will be collected at baseline. VAS and EQ-5D-5L questionnaires will be conducted on completion of treatment and at each follow-up point (see below).
Blood samples and storage
If taken as part of routine clinical care, the patient’s baseline blood tests (haemoglobin, white cell count, platelet count, electrolytes, liver function tests and clotting tests) will be recorded. Patients will be consented to have an additional blood test for highly sensitive C reactive protein, which will be sent to the ORTU for analysis. This blood will be centrifuged, labelled and stored as part of the ORTU Collection of the Oxford Radcliffe Biobank. Genetic compositional analysis may also be undertaken on participants’ samples if specific consent for this has been obtained.
Digital air leak measurement
All patients requiring pleural intervention will have digital measurement of air leak using a digital measuring device (Thopaz+, Medela, Switzerland). Measurements will be taken immediately postintervention (day 0) for the ambulatory device or chest drain, then daily at around the same time on days 1–4 (or until chest drain/device removal). On each occasion the Thopaz+ (Medela) device should be attached to the chest drainage device (either ambulatory device or standard chest drain, dependent on the arm of the trial) for 10 min, with the device set to −0.4 kPa (ie, providing no suction but as close to normal physiological pleural pressure as possible). During this time, the air leak measurement should be recorded by reading the measurement at 1, 5 and 10 min. These data will be uploaded to OpenClinica.
Failure of medical treatment
There is no robust evidence on the ideal timing for surgical intervention. Current BTS guidelines suggest that cases of persistent air leak or non-re-expansion should be referred after 3–5 days.7 To achieve objective outcomes for this study, the following measurable and documentable criteria have been developed to ensure consistent practice and will be recorded as part of the study in all cases. Referral for thoracic surgery will occur in the presence of all of the following:
Day 4 postinsertion of chest drain, persistent air leak as measured by ‘bubbling’ chest drain attached to underwater seal, or evidence of ongoing air leak through an ambulatory device.
Persistent pneumothorax on CXR.
Patient agreement.
No contraindication to thoracic surgery.
In all cases, the clinical parameters and radiology (both those referred to surgery and those successfully treated with medical management) will be blindly assessed at the end of the study to ensure the discharge and surgery criteria were robustly followed.
CT scan
CT scan will be conducted in all patients (approximately 2–4 weeks after enrolment) to detect lung parenchymal abnormalities. These should be conducted as per protocol (see online supplementary appendix C), including high-resolution limited cuts apically. Any patient who has a CT as part of their clinical care will not be required to have an additional scan. The CT regimen was designed to be low dose (total 3.4 mSv, equivalent of 1-year background radiation). The benefit of CT scanning in every young patient with pneumothorax is not proven and controversial due to the additional radiation dose. This trial aims to determine whether CT findings can predict long-term outcomes and therefore be of clinical utility.
Trial follow-up
All patients will be followed up 1 week after completion of treatment, then at 1, 6 and 12 months from enrolment. At each time point, the patient will complete the VAS and EQ-5D-5L questionnaires, have a CXR, and documentation of smoking status and any recurrence of pneumothorax. Every effort should be made to ensure patients attend their follow-up appointments. However, patients not able to attend may be contacted by phone to check for complications or recurrence. If this is not possible, then data on recurrence can be collected through medical notes.
Primary outcome
The primary outcome measure will be total length of stay in hospital to include primary hospital stay and readmissions up to 30 days postrandomisation. Patients remaining in hospital overnight will be classed as 1 day; those discharged on the same day (after either successful NA (needle aspiration) treatment or treatment with pleural vent) will have a zero length of stay. These attendances will be captured in the health economic analyses. Thirty days has been chosen on the basis of previous data suggesting that the majority of conservatively treated (non-surgical) air leaks will have resolved within 14 days of initial treatment in pneumothorax, and a 30-day outcome point is therefore conservative and will reliably capture all related readmissions. Readmission will be defined as the requirement of emergency (non-planned) visit to hospital requiring any form of contact with medical services (not restricted to further pleural interventions) in relation to the pneumothorax. This will not include planned day case reviews for the outpatient treated population.
Secondary outcomes
The following are the secondary research questions:
Is ambulatory care and early discharge safe and cost-effective?
Is patient experience improved with an ambulatory device, including pain of procedure, breathlessness, quality of life assessments (EQ-5D-5L) and time to return to working status?
What is the recurrence rate of pneumothorax over 6 and 12 months?
Does radiological evidence (on CT scanning) of emphysema-like changes and inflammation predict long-term outcome (ie, recurrence of pneumothorax within 12 months)?
Can digitally measured air leak and its evolution over treatment predict short-term clinical trajectory in patients with pneumothorax, including failure of treatment (ie, prolonged air leak or non-expanded lung)?
Sample size calculation
The sample size for the primary outcome was determined to detect a difference in hospital stay of 2.3 days: from a mean of 4 days admission in the control arm to an expected mean of 1.7 days in the intervention arm (SD in both groups 6.0). These means include the calculation that up to 50% of the control arm may be successfully treated with aspiration alone (ie, zero-day admission). It is assumed, conservatively, that ~20% of patients in the intervention arm will require a readmission. Therefore, to detect this difference, accounting for non-parametric data requires 236 patients in total, including a 10% attrition rate, an 80% power and a 5% two-sided significance level. Our previous pleural studies have demonstrated an attrition rate for the primary outcome measure of <5%.
End of trial
The trial will end once 236 patients have been randomised and all patients have completed 12 months of trial follow-up.
Statistical analysis plan
The primary outcome will be analysed using the Mann-Whitney U test. The median hospital stay (expected to be non-normally distributed) will be reported for each arm, and the 95% CI for difference in medians will be calculated using an exact test.
As a sensitivity analysis, survival analysis techniques will be used. Kaplan-Meier survival curves will be presented graphically. Survival (ie, time without recurrence of pneumothorax and in follow-up) will be compared between arms using the Gehan-Breslow-Wilcoxon test, which is more appropriate than the log-rank test when the rate of early events is high. Cox proportional hazards regression will also be used to calculate the HR and 95% CI.
Continuous secondary outcome measures will be analysed using analysis of covariance adjusting for baseline score. Results will be reported as adjusted mean difference between treatment arms, with 95% CI and p values. Categorical secondary outcome measures will be analysed using the χ2 test. Time to event secondary outcome measures will be analysed in the same way as the time to event analysis of the primary outcome measure. Patient-related factors and time to return to work will be collated blindly and verified by a central panel, unaware of interventional arm and/or requirement of surgery.
A Consolidated Standards of Reporting Trials diagram will be presented, including the number of patients screened for the study, the numbers randomised, the numbers receiving the interventions, the numbers lost to follow-up and excluded (with reasons), and the number of patients included in the primary analysis.
Safety reporting
Data will be collected at each patient’s trial visit regarding any serious adverse events (SAE; as defined by good clinical practice guidelines). All SAEs causally related to trial interventions will be reported to the sponsor and to the relevant oversight bodies, and will be followed until they resolve or stabilise.
SAEs in this study which should be reported immediately (ie, within 24 hours) to ORTU include tension pneumothorax occurring during treatment (until discharge), blockage of drain with clinical consequences (eg, patient unwell, further procedure), major haemorrhage which requires specific intervention (eg, blood transfusion) and any additional emergency pleural procedure as deemed necessary by the responsible local physician (eg, large-bore chest drain insertion).
The following are considered to be expected AEs associated with the proposed trial interventions for this trial:
Pain at drain site.
Minor haemorrhage not requiring specific intervention such as blood transfusion or surgery.
Subcutaneous emphysema.
Pleural infection.
Unintentional removal (‘falling out’).
Recurrence of pneumothorax/worsening of ongoing pneumothorax (if no evidence of initial full resolution).
Re-expansion pulmonary oedema.
Any further (non-emergency) pleural procedure required.
Recurrence of pneumothorax is expected in approximately 33% of PSP within 1 year. This is defined as a recurrence of pneumothorax (if fully resolved and documented as fully resolved) or worsening of ongoing pneumothorax (if discharged but no evidence of full resolution). These do not need to be reported as an SAE (even if they meet the criteria of requiring hospitalisation or prolongation of their hospital stay), as all the information will be explicitly captured at the follow-up visit.
Cost-effectiveness analysis
The perspective adopted in the economic analysis will be that of the UK National Health Service. As a result data will be collected on hospital stay, outpatient contact (primary and secondary care) and resource usage.
As the main outcome measure in the economic evaluation will be incremental cost per quality-adjusted life year (QALY) gained, generic quality of life information will be collected. The EuroQol EQ-5D-5 levels—a widely used generic multiattribute utility scale—will be completed for each patient at baseline and at 1-week, 1-month and 6-month assessments to measure patients’ general health-related quality of life. For QALY construction, EQ-5D-5L results will be translated into utility values using published UK population valuations.
A within-trial cost-utility analysis will explore the incremental cost per QALY gained of ambulatory care when compared with standard care. Cost and effect results will be reported as means with SD, with mean differences between the two patient groups reported alongside 95% CIs. Cost-effectiveness and health economic analyses may be published in a separate publication to the primary trial results.
Safety monitoring and interim analyses
The Data Safety Monitoring Committee (DSMC) consists of independent experts (medical experts and a statistician) external to a trial who assess the progress, conduct, participant safety and critical endpoints of a clinical trial. A blinded interim analysis of the primary outcome (hospital stay) will be undertaken after approximately 50% of patients have been recruited in order to assess the assumptions made in the sample size calculation, and not to directly compare the intervention arms. No correction of the significance level of the final analysis is planned on this single assessment of early event rate by the DSMC.
Changes to the protocol after trial commencement
The trial details documented here are consistent with the RAMPP trial protocol V.8.0 (date: 3 November 2017). A summary of the trial amendments can be found in online supplementary appendix D. Major amendments included reducing the inclusion age to ≥16 years old, clarification that patients enrolled in the observational cohort study could subsequently be enrolled in the treatment part of the study if they require intervention, and clarification of pneumothorax recurrence and re-expansion pulmonary oedema as expected AEs.
Publication policy
The trial results will be presented at regional, national and international conferences and scientific meetings with publication in a peer-reviewed journal (authorship will be according to the journal’s guidelines). A lay summary of the study results will be circulated to consented patients and relevant funding bodies.
Footnotes
Contributors The protocol was drafted by RH, ML-S and NR. The final protocol and manuscript were reviewed and approved by all authors.
Funding This work was funded by NIHR Research for Patient Benefit (grant number PB-PG-0213-30098). The ambulatory devices (Pleural Vent) and consumables were supplied by Rocket Medical (UK). The digital suction devices (Thopaz+) and consumables were supplied by Medela (Switzerland).
Competing interests NR has received consultancy fees from Rocket Medical. All other authors have no competing interests to declare.
Patient consent for publication Not required.
Ethics approval The trial is being conducted in accordance with the Declaration of Helsinki and Good Clinical Practice.
Provenance and peer review Not commissioned; externally peer reviewed.