Article Text

Abstract
Background Human enterovirus D68 (EV-D68) was first isolated in 1962 and has aroused public concern recently because of a nationwide outbreak among children in 2014–2015 in the USA. The symptoms include fever, runny nose, sneezing, cough and muscle pains. It might be associated with severe respiratory illness in individuals with pre-existing respiratory conditions and its potential association with acute flaccid myelitis is under investigation. In Asia, EV-D68 cases have been reported in several countries.
The study We aimed to understand the EV-D68 prevalence and their genetic diversity in Hong Kong children.
Methods A total of 10 695 nasopharyngeal aspirate (NPA) samples from hospitalised patients aged <18 years were collected from September 2014 to December 2015 in two regional hospitals. NPAs tested positive for enterovirus/rhinovirus (EV/RV) were selected for genotyping. For those identified as EV-D68, their complete coding sequences (CDSs) were obtained by Sanger sequencing. A maximum-likelihood phylogeny was constructed using all EV-D68 complete coding sequences available in GenBank (n=482).
Results 2662/10 695 (24.9%) were tested positive with EV/RV and 882/2662 (33.1%) were selected randomly and subjected to molecular classification. EV-D68 was detected in 15 (1.70%) samples from patients with clinical presentations ranging from wheezing to pneumonia and belonged to subclade B3. Eight CDSs were successfully obtained. A total of 10 amino acid residue polymorphisms were detected in the viral capsid proteins, proteases, ATPase and RNA polymerase.
Conclusion B3 subclade was the only subclade found locally. Surveillance of EV-D68 raises public awareness and provides the information to determine the most relevant genotypes for vaccine development.
- clinical epidemiology
- viral infection
- respiratory infection
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
On the 2014 outbreak of enterovirus D68 (EV-D68) in the USA with severe respiratory diseases in children and its possible association with acute flaccid myelitis, we would like to examine if the EV-D68 detected in children in Hong Kong has been evolving.
We described the molecular epidemiology of EV-D68 in Hong Kong and identified a 1.70% EV-D68 positive rate in hospitalised paediatric patients tested positive with enterovirus with all of their taxonomic classifications of subclade B3.
This piece of research would provide the genomic data of EV-D68 strains isolated from hospitalised children in Hong Kong between September 2015 and December 2016. This would be useful for researchers in the field to apply these data to their evolutionary analysis in East Asia.
Introduction
Enterovirus D68 (EV-D68) was first isolated from four children with bronchiolitis and pneumonia in 1962 in California. EV-D68 is a single-stranded positive-sense RNA virus that belongs to the family Picornaviridae in the genus Enterovirus, a member of D species. The genome of EV-D68 consists of approximately 7.2 kb nucleotides with a single open reading frame (ORF), from which several capsid proteins, protease (Pro) and RNA-dependent RNA polymerase are translated (ie, 5′-viral protein (VP) 4, VP2, VP3, VP1, 2APro, 2B, 2C, 3A, 3B, 3CPro, 3DPol-3′).
In 2014, an unprecedented EV-D68 outbreak started and more than 2000 cases were reported in 20 countries with more than half of the cases (n=1153) reported in the USA across 49 states,1 the Americas,2 Europe3 4 and Asia.5 EV-D68 causes a wide range of respiratory symptoms in children, and the disease spectrum ranges from asymptomatic to severe respiratory complications. A study showed that cough, wheezing and chest indrawing are three of the most common manifestations.6 For non-respiratory symptoms, fever, vomiting and malaise are usually observed. In addition, EV-D68 has been reported to be associated with neurological features (eg, acute flaccid paralysis) in children and adults.7–11
Since the 2014 outbreak, investigations have been done globally on the genetic variability of EV-D68 strains among different geographical localities. Phylogenetic analysis was used to assign the strains to a specific clade.12 Up until now, EV-D68 were assigned into four clades (A, B, C and D) by maximum-likelihood (ML) algorithm based on the VP1 region.12–14 With the increasing molecular epidemiology data acquired globally, subclades of B and D were proposed including B1,7 B2,13 B3,15 D1 and D2,7 16–18 while B3 strains seem to be dominating in recent reports in Japan,19 20 Sweden21 and Italy.22
Apart from assigning virus strains into individual clades and subclades, molecular analysis of viral protein mutation is another important aspect to understand the virulence of the virus. The mutations in VP1 is of special interest in the field as it is responsible for the host receptor binding and contains serotype-specific neutralisation sites.23 In the 2014 North America outbreak, a persistent evolution in VP1 region was reported.14 The identification of the two deletion blocks at the 5′ untranslated region of the viral genome nucleotide positions 681–704 (24 nt) and 721–731 (11 nt), located between the end of internal ribosome entry site and the polyprotein ORF, was also found to be an important mutation as it would affect the initiation of viral translation and enhance the translation efficiency.12 20 Therefore, we aim to provide information of the mutations identified in the EV-D68 detected in this study in order to assess if new virulent factors would be identified.
In Hong Kong, an earlier study screening through 6800 nasopharyngeal aspirates (NPAs) collected from both children and adults in 2010–2014 was conducted with 30 (0.44%) of them being EV-D68 positive. The VP1 sequences from nine of these samples were analysed and they belonged to A1, A2, B1 and B3 subclades.16
In this study, we covered the subsequent 2 years of the previous study, focusing on the EV-D68 disease burden in hospitalised children under 18 years old in Hong Kong. We detected a cluster of EV-D68 cases in the winter of 2015. The clinical outcomes were documented, and we completed the genome sequencing of these strains. A comprehensive phylogenetic tree was constructed using all the EV-D68 strains with complete CDS nucleotide sequences available from global studies.
Methods
Clinical specimens and patient information
Prince of Wales Hospital (PWH) and Alice Ho Miu Ling Nethersole Hospital (AHN) are two regional hospitals in the East Cluster of the New Territories in Hong Kong with a catchment of 1.5 million population. NPAs were collected from hospitalised children from these two hospitals from September 2014 to December 2015 (table 1). NPA would generally be collected from a paediatric patient with respiratory symptoms or with fever without respiratory symptoms.
Number of NPA specimens tested by multiplex real-time PCR assay during period from Sep 2014 to Dec 2015 in patients less than 18 years old
The respiratory pathogen screening using NPA specimen is part of the routine clinical diagnosis in the public hospital in Hong Kong and it is clinician directed. The presence of EV/RV was tested prospectively by the multiplex real-time PCR as part of the diagnostic work-up, while the enterovirus genotyping was performed retrospectively for this project.
Two NPAs collected from hospitalised patients under 18 years old which were positive with EV/RV and without detectable co-infection were selected randomly per calendar day. The samples obtained were frozen at −80°C for enterovirus genotyping.
The demographic information, clinical features, laboratory results and outcome of illness of the EV-D68-infected patients were obtained from the electronic clinical record and analysed by two paediatricians (CYL and KLH). This study did not set any exclusion criterion.
RNA extraction, RT-PCR and enterovirus genotyping by VP4/2 region sequencing
Fifty microlitres of the NPA was used for viral RNA extraction using TaKaRa MiniBEST viral RNA/DNA extraction kit V.5.0 (TaKaRa, Japan). Thirty microlitres of RNase-free water was used to elute the RNA. Then 6.5 µL eluent was used as template for reverse-transcription PCR (RT-PCR). RT-PCR was performed using TaKaRa PrimeScript RT reagent kit (Perfect Real Time) according to the manufacturer’s instruction.
Nested PCR was adopted to amplify the VP4/2 region.24 The primers used are listed in online supplementary table S1. VP4/2 nucleotide sequence was obtained by Sanger sequencing. Basic Local Alignment Search Tool (BLAST)25 command line tool was used to perform taxonomy assignment. The BLAST database used for classification comprised VP4/2 nucleotide sequences of all confirmed genotypes under species Rhinovirus A, B, C and Enterovirus D. VP4/2 sequencing data were compared with reference sequences in the database by BLAST.26
Supplemental material
Complete coding sequence (CDS) sequencing
EV-D68-positive samples identified in the VP4/2 sequencing underwent CDS sequencing. Twelve pairs of primers were designed (online supplementary table S3) to cover the full genome of EV-D68 with 200–300 bp long overlapping region between adjacent regions. PCR was performed using TaKaRa Taq HS Perfect Mix kit (TaKaRa, Japan) and the PCR products underwent two-direction Sanger nucleotide sequencing.27 Assembly of the sequencing fragments representing the full EV-D68 genome was done manually using multiple sequences alignment function built in SnapGene software (from GSL Biotech; available at www.snapgene.com).
Supplemental material
Data collection and compilation of EV-D68 complete CDS data pool
All EV-D68 nucleotide sequences with complete or nearly completed CDS that were available in GenBank (as of March 2018) were collected. Entries without complete CDS nucleotide sequence (ie, the length of CDS region, located from 733 to 7299) based on the co-ordinator of Fermon strain ( GenBank accession no. AY426531) were discarded. A total of 482 sequences were distributed to eight datasets corresponding to clades or subclades prototype (n=2), A (n=38), D1 (n=2), D2 (n=8), C (n=48), B1 (n=277), B2 (n=26) and B3 (n=70), which were determined by ML phylogenetic analysis using RAxML V.8.28
Sequence alignment, phylogeny, pairwise distance and amino acid polymorphism
Sequence alignment was carried out using MAFFT software29 through CIPRES scientific gateway.30 ML phylogenetic trees were constructed using RAxML V.828 under the GTR model31 with the gamma distribution to model site heterogeneity (GTRGAMMA), using 1000 bootstrap replicates. The EV-D68 prototype strain (ie, Fermon, accession no. AY426531) was included as an outgroup.32 The tree was drawn to scale, with branch lengths measured in the number of substitutions per site. Phylogenetic trees were visualised with ggtree package33 in R34 and iTOL V.4.0.3.35
Pairwise distances (ie, the number of base differences per site from between sequences) were calculated in MEGA6.36 The CDS and VP1 sequences were separated into different groups (ie, prototype, A, D1, D2, C, B1, B2, B3) based on the phylogenetic clades/subclades. Inter-clade, intra-clade, inter-subclade and intra-subclade mean pairwise distances were calculated, and the strain list included in each group could be found in online supplementary table S2.
Supplemental material
Amino acid alignment was carried out by Clustal W program37 implemented in MEGA6.36
Statistical test
Within the EV/RV-positive patients under 18 years old, we further stratified them into two age groups, preschool age (<5 years) and school age (6–17 years), respectively. Children in these two age groups were in different developmental stages and have different exposure in their environments, and their susceptibility towards EV/RV infections might differ accordingly. Due to the limited number of EV-D68 detected, we did not break down to finer age group. Chi-square test was used to determine the significance level of difference between the EV-D68 positive rate among different age groups. A p value of <0.05 would deem as significant. All statistical analyses were carried out using R.34
Nucleotide sequence accession number
The nucleotide sequences of those 15 EV-D68 strains detected in our study are available in GenBank under accession numbers MG739632–MG739646.
Patient and public involvement
This research was done without patient involvement. Patients were not invited to comment on the study design and were not consulted to develop patient-relevant outcomes or interpret the results. Patients were not invited to contribute to the writing or editing of this document for readability or accuracy.
Results
Detection of enterovirus/rhinovirus (EV/RV) from September 2014 to December 2015
From September 2014 to December 2015, a total of 29 211 NPA specimens were collected from hospitalised patients of all age groups in which 36.61% (n=10 695) were from patients under 18 years old. A total of 2662/10 695 (24.89%) NPAs were identified as EV/RV positive.
Preschool-age children had a higher overall EV/RV positivity than school-age children (table 1, figure 1, p<0.0001), while their monthly distributions were similar. Peaks were observed around May and October.
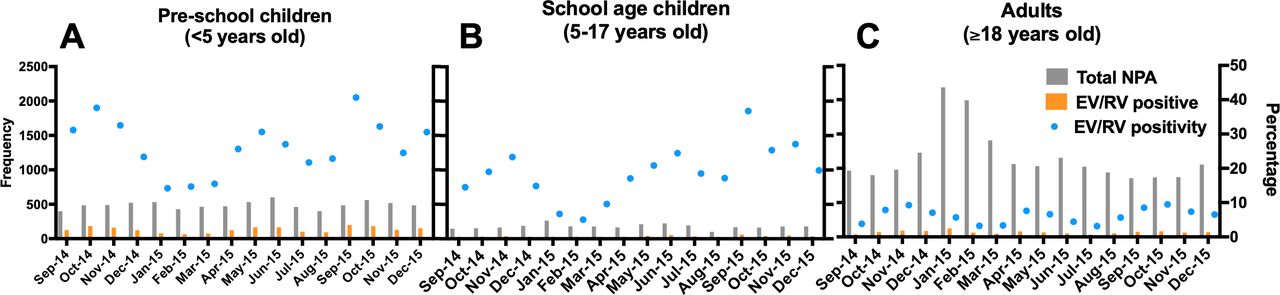
Monthly distribution of EV/RV-positive NPA specimens detected in hospitalised (A) preschool-age children (0–5 years), (B) school-age children (5–17 years) and (C) adults (above 17 years) in Prince of Wales Hospital and Alice Ho Miu Ling Nethersole Hospital from September 2014 to December 2015. A total of 29 211 NPA samples were collected in the two regional acute government hospitals in the East Cluster of New Territories in Hong Kong serving approximately 1.5 million people. These samples were sent for virological tests. The frequency of the number of NPAs collected (grey) and the EV/RV-positive samples (orange) were plotted against the left y-axis. EV/RV-positive sample of individual age group was calculated for each month (blue). EV/RV, enterovirus/rhinovirus; NPA, nasopharyngeal aspirate.
Furthermore, the positive rate of EV/RV in adults (ie, >18 years old) was 5.91%, which was significantly lower than those in preschool-age children (ie, 0–5 years) (27.08%, p<0.00001) and school-age (ie, 6–18 years) children (18.91%, p<0.00001) (figure 1).
Detection of EV-D68 by VP4/2-targeted PCR and Sanger sequencing
A total of 882/2662 (33.13%) NPAs from patients aged under 18 years and tested positive with EV/RV were selected randomly to undergo molecular classification. A total of 15 specimens (1.70 %) were detected with EV-D68 (table 1). Seven of them came from AHN and the remaining eight samples were collected in PWH. They were collected in October (n=6, 3.37%), November (n=3, 5.17%) and December (n=6, 10.71%) in 2015. To explore if the detection of EV-D68 in the last quarter of 2015 was epidemiologically linked, four more EV/RV-positive samples per week were extracted from April to October in 2015 for the viral genotyping; nevertheless, no extra EV-D68 specimen was identified. Due to limitation of NPA availability, further exploration could not be done in the remaining months (ie, September–December 2014, January–March 2015 and November–December 2015).
Demographic and clinical information of patients with EV-D68 infections
As we did not include adult NPA specimens in this study, all 15 EV-D68-positive patients were under 18 years old (median age 4 years, range from 5 months to 15 years; table 2). Twelve were male and three were female with six of them presented with asthma attack. Besides pneumonia and bronchiolitis, one of the patients was suffering from Kawasaki disease at the time when EV-D68 was detected.
Clinical characteristics of the 15 cases of EV-D68 infection
Genome features and pairwise distance analysis between strains from different clades and subclades
Seven complete CDSs (cases 1–6 and 8) and one nearly completed CDS (case 7) were successfully acquired in the present study with the mean GC content of 41.83%. The complete nucleotide sequences of the EV-D68 strains showed 0% to 2.9% differences to each other, and the complete amino acid sequences (2188 aa) showed 0% to 0.8% differences to each other.
The pairwise distance calculation based on 482 global EV-D68 strains indicated that strains within B3 subclade had a mean p-distance of 0.017 (ranged from 0 to 0.035) and 0.018 (ranged from 0 to 0.036) for CDS nucleotide sequence and VP1 region, respectively (table 3).
Pairwise distances of CDS and VP1 nucleotide sequences between clades and subclades of global EV-D68 strains*
Maximum likelihood phylogeny of local and global strains
Global EV-D68 sequences with a complete CDS sequence available (n=482, as of March 2018) including seven of complete CDSs (cases 1–6 and 8) acquired in this study were included in the phylogenetic analysis. All seven strains belonged to subclade B3 (figure 2 and online supplementary table S2), which were closely related to strains isolated in Shenzhen, China (n=3) in 2015; Osaka, Japan (n=5) in 2015; and New York, USA (n>20) in 2016. For the strains that we were not able to complete the CDS (ie, cases 7, 9, 10–15), a further phylogenetic analysis of the VP4/2 region indicated that all of them came from B3 subclade (data not shown).

Phylogenetic trees of complete coding sequences (CDSs) of all EV-D68 strains deposited in GenBank database. The tree was rooted with the EV-D68 prototype Fermon strain ( GenBank: AY426531). A total of 482 strains (including seven complete CDSs obtained in this study) were included in this analysis, and their details are shown in online supplementary table S2. Positions 733–7299 (ie, complete CDS region) were used as the alignment region in the analysis. The outer ring indicated the year of virus isolation and encoded in the gradient of red. The inner ring indicated the geographical origin (GEO) of the viral strains. The colour coding within the phylogenetic tree was marked according to the clade A in green, B1 subclade in purple, B2 subclade in orange, B3 subclade in moss green, clade C in blue and clade D in pink.
An enlarged phylogenetic tree of B3 subclade that comprised 70 strains collected from East Asia (n=42) and North America (n=28) (figure 3) indicated that Hong Kong/CUHK47134 (case 5), Hong Kong/CUHK61194 (case 6) and Hong Kong/CUHK12619 (case 2) grouped with strains from Osaka, Japan in 2015 (ie, A250-OsakaC-JPN-2015, A244-OsakaC-JPN-2015, A241-OsakaC-JPN-2015 and A252-OsakaC-JPN-2015) with the overall mean similarity of their complete CDS of 99.68%. Besides, Hong Kong/CUHK16709 (case 4), Hong Kong/CUHK09970 (case 1), Hong Kong/CUHK15980 (case 3) and Hong Kong/CUHK66649 (case 8) were closely related to the EVD68/SZ01/CHN/2015 strain detected in Shenzhen, China in 2015, and the overall mean similarity of their CDS nucleotide sequence was 99.91%.

{kind=link}
{kind=link}
{kind=link}
Enlarged phylogenetic tree of EV-D68 subclade B3 strains based on coding sequence (CDS). The branches were coloured according to GEO information. The EV-D68 strains with CDS obtained in this study (case 7 was excluded as it was not a complete CDS) are highlighted with a pink background. These seven strains clustered with those collected in neighbour geographical location, such as ‘A250-OsakaC-JPN-2015’ from Osaka, Japan and ‘EVD68/SZ01/CHN/2015’ from Shenzhen, China. CDS, coding sequence.
Amino acid residue polymorphisms of the eight complete EV-D68 genomes acquired in this study
Ten amino acid polymorphisms were detected among the eight genomes with their CDS aligned, namely Q44H in VP4, A220T in VP2, F532I in VP3, S694G in VP1, A965T in 2A, I1227N in 2C, I1445V and H1484Q in 3A, and F1973S and V2080A in 3D (table 4). Furthermore, two more aa residue polymorphisms, I470V in VP3 region and S695G in VP1 region, were identified by aligning these eight CDSs to the two representatives of B3 clade, strain ‘A250-OsakaC-JPN-2015’ (GenBank: LC107900.1) and ‘EVD68/SZ01/CHN/2015’ (GenBank: KU982558). Of note, neither of those two VP1 region residues (S694G and S695G) mentioned above located in the BC-loop (residues 89–106) and DE-loop (residues 140–152) which were thought to be important in both host–receptor binding and viral antigenicity.38
Amino acid residue polymorphisms detected in the EV-D68 CDS obtained in this study
Discussion
We reported the EV-D68 epidemiology data in hospitalised children in Hong Kong from September 2014 to December 2015. According to the respiratory pathogen screening records, children had a much higher EV/RV infection rate than that of adults. The genotyping of enterovirus performed for 33% of the paediatric cases resulted in the identification of 15 EV-D68 cases, and fortunately, none of them led to fatality. Detailed molecular classification of the VP4/VP2 sequences of these 15 strains indicated that all of them are of B3 subclade. Seven strains of EV-D68 had their complete genome sequenced. In the ML phylogenetic tree analysis, the strains identified in the current study clustered close together with strains isolated in Shenzhen, China (n=3) in 2015 and in Osaka, Japan in 2015. Ten amino acid polymorphisms were detected among the eight sequenced CDSs in this study. These sites distribute along the viral genome and were not reported in the previous study. Whether these changes would be responsible for the virus virulence would rely on more surveillance work in the future.
We have our study focused on paediatric population because of the increased EV-D68 seroprevalence observed by Kamau et al in infants and young children in UK from 2006 to 2016.39 The acquisition of the capacity for a subset of the contemporary EV-D68 strain in the 2014 outbreak to enter and replicate in human neuronal cells (SH-SY5Y cell model)40 inferred the expansion in tropism for neural system cells, which might be associated with neuropathology, such as acute flaccid myelitis. The differential pathological outcomes seem to be associated with the strains in subclade B1 which carries 12 substitutions as of those presented in paralysis-causing enterovirus.41 With the continuous evolution and recombination of EV-D68, novel subclade emerged which warrants additional surveillance.
According to the routine respiratory virus screening data in samples collected from hospitalised patients, children had a significantly higher EV/RV positive rate than adults. Both groups have similar seasonality with the peak seasons during May–June and September–October within the investigation period (table 1). Intriguingly, all the EV-D68 strains detected in our study emerged in winter months (October, November, December). We explored more NPA samples by expanding the number of samples (only for those collected in April–October 2015); no further EV-D68 were identified. In another study from Hong Kong, EV-D68 peaks could be observed in both late spring/summer (May to August) and in late autumn/early winter (October to December in 2011).42 More future surveillance works will be critical for disentangling EV-D68 seasonality in Hong Kong.
Previous review showed that asthma was the most common underlying pulmonary disease among EV-D68-positive patients.6 Within the 15 EV-D68-positive patients identified in our study, 40% (6/15) of them were diagnosed with asthma attack by clinicians according to their clinical record. Notably, the prevalence of asthma varied between different reports. In the reports from Chicago and Kansas in 2014, 73% (8/11) and 53% (13/19) of children positive with EV-D68 infection had a history of asthma or wheezing. Besides, a study from Japan also indicated that EV-D68 was detected in 74.3% (26/35) of hospitalised children who were suffering from asthma attacks in 2010.43
In terms of the viral genome diversity, the sequence alignment and phylogenetic analysis revealed that all the 15 strains sequenced in this study belonged to B3 subclade. The EV-D68 strains circulating in 2015 in Hong Kong16 42 and its neighbouring region, Shenzhen,44 Taiwan district15 and Osaka45 have high similarity. Most of the EV-D68 strains identified in this region belong to B3 subclade (figure 2 and online supplementary table S2). In contrast to the B1 strains that caused an outbreak in the USA in 2014, recent studies showed that an increasing number of B3 subclade were found to be circulating in America and Europe. An outbreak of B3 subclade was reported in Stockholm in 2016,21 and it formed a tight cluster together with the B3 strains identified in the USA in 2016.46 Furthermore, eight adults and 14 children were identified to be infected by EV-D68 of B3 subclade in the Netherlands in June and July 2016.47
Twelve single amino acid residue polymorphisms were identified when aligning the current strains with the B3 representative strains. The complete CDSs which are available in GenBank would allow the investigation of their biological relevance in a large scale of sequence analysis with their association to clinical manifestation which may help in vaccine development.48 The causal relationship of these deletions and mutations to the increased virus virulence would require evidence from biological study, such as the infection experiment performed by Hixon et al in neonatal mice, which indicated the viral infection and killing of spinal cord motor neurons.49
In conclusion, we reported the epidemiology data of EV/RV and 1.7% of the EV/RV-positive NPA specimens isolated from in children in Hong Kong were positive with EV-D68. All the 15 strains came from B3 subclade. Deletion blocks and SNPs discovered in our study could help to study epidemiological and evolutionary features of EV-D68 in Hong Kong and its neighbouring regions.
Limitations
The exclusion of co-infection might result in the unavailability to assess the influence of EV-D68 in leading to other complications. The retrospective nature of this study could not allow us to collect standardised clinical data for a comprehensive analysis. Due to limited resources, our study design was limited to random selection of EV/RV-positive specimens to undergo further EV genotyping which was only 33% of our overall EV/RV specimens. Furthermore, due to the limitation in specimen availability, the expanded detection could only be performed in NPAs collected from April to October 2015. This might limit the full understanding of EV-D68 distribution in the accessible NPA specimens. Besides, the samples used in this study were coming from the PWH and AHN, the two regional hospitals located in the East of the New Territories of Hong Kong with a population caption of 1.5 million, and the coverage of investigation would be enhanced if this is a territory-wide study.
Acknowledgments
Miss Tiffany T J Li, Mr Anthony Kwok and Miss Hailey Y Tsui are thanked for their help in RNA extraction. Dr Joseph G S Tsun is thanked for his technical assistance and Professor Haitian Wang for the statistical advice.
References
Footnotes
Contributors HW and WYRC designed the study. KSPC and CMAY conducted virological investigations. CYL, KLH and T-FL provided clinical data. KT and HW conducted experiments. HW and ZC performed data analysis. HW, KT and WYRC wrote the manuscript. KT, T-FL, KLH, KSPC and WYRC performed critical review of the manuscript.
Funding This work was supported by the RGC/ECS 24107017 and Direct Grant 2015.1.055 to WYRC, 2014.1.063 to T-FL and the one-off research grant for the CUHK–UMC Utrecht Joint Research Laboratory of Respiratory Virus and Immunobiology from the Research Committee of the Chinese University of Hong Kong.
Disclaimer The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was approved by the Joint Chinese University of Hong Kong–New Territories East Cluster Clinical Research Ethics Committee (CREC no. 2015.612).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open-access repository.