Article Text

Statistics from Altmetric.com
Key messages
What is the key question?
How beneficial is reslizumab in patients with severe refractory asthma?
What is the bottom line?
Reslizumab provides significant clinical benefit and is well tolerated in patients with refractory asthma and elevated blood eosinophil counts.
Why read on?
This post hoc analysis provides the first evidence that reslizumab resulted in significant benefit in exacerbations, lung function, asthma control and symptoms, and quality of life for patients with severe refractory asthma and elevated blood eosinophil levels, which was consistent with prior studies in patients with asthma and eosinophilia.
Introduction
Asthma is a common disease, affecting an estimated 334 million people worldwide, with considerable impact on quality of life and high associated costs.1–3 Asthma severity is assessed retrospectively from the level of treatment required to control symptoms and exacerbations. Approximately 5%–10% of patients with asthma are believed to suffer from severe disease.4 Patients with severe asthma typically require ongoing maintenance therapy with high-dose inhaled corticosteroid (ICS)/long-acting beta-agonist (LABA).2 Furthermore, systemic corticosteroids (SCS) are often required for potentially life-threatening exacerbations, but are associated with long-term risks of severe side effects.5
Patients with severe asthma which is uncontrolled despite optimal therapy, good compliance, trigger avoidance and management of comorbidities are classified as having refractory asthma (RA), associated with persistent symptoms despite maximal therapy and extensive re-evaluation of asthma management.2 6 7 Up to 3.6% of patients are estimated to suffer from severe RA despite high medication use.6 8
Eosinophils are instrumental in the pathogenesis of asthmatic airway inflammation: their numbers have been correlated to lung function impairment9 and increased risk of clinical asthma exacerbation (CAE).2 6 10 One phenotype of severe asthma is characterised by persistent airway inflammation with eosinophils.11 The eosinophil viability-enhancing factor, interleukin-5 (IL-5), controls their differentiation and maturation within the bone marrow and stimulates migration to sites of inflammation by acting on the eosinophil’s IL-5 receptor.12 Patients with this phenotype often require SCS, with their increased risk of adverse events (AEs).13 There is, therefore, a significant need for improved treatment options that target eosinophils for patients with RA and eosinophilia.
Reslizumab is an IgG4 kappa, humanised, monoclonal antibody that targets IL-5, reducing eosinophil proliferation and decreasing levels of airway inflammation.14 15 Randomised, placebo-controlled, phase 3 trials have shown reslizumab to reduce CAE rates, and improve lung function and patient-reported outcomes in inadequately controlled eosinophilic asthma.16–18 Specifically, in two duplicate 52-week trials, reslizumab reduced the rate of CAEs by 54% compared with placebo.17 Reslizumab demonstrated an overall similar safety profile to placebo. Three patients in the reslizumab group had anaphylactic reactions that were deemed by the investigators to be treatment-related; all responded to standard treatment and were discontinued from the study.17–19
Patients with RA who pose a particular challenge in the management of severe asthma were included in these studies. Clinical experience with anti-IL-5 treatments indicate that patients with severe RA might benefit particularly from these targeted treatments, suggesting that the responses of this subgroup to the effects of reslizumab should be analysed separately. Here, we present a subgroup analysis of data from two duplicate 52-week trials to specifically assess reslizumab as a treatment option for patients with RA. Our hypothesis was that patients in this subpopulation would achieve clinically relevant improvements in asthma outcome measures with reslizumab compared with placebo.
Methods
Study design and patients
This study was a post-hoc analysis of a subpopulation of patients with RA from two previously published multi-centre, double-blind, randomised, placebo-controlled phase 3 trials (NCT01287039 and NCT01285323). The design and methodology of these trials have been previously published, and are described only briefly here.17
Patients aged 12–75 years with a current blood eosinophil count of ≥400/µL, inadequately controlled asthma (baseline Asthma Control Questionnaire (ACQ) score ≥1.5), forced expiratory volume in 1 s (FEV1) reversibility of ≥12% to β-agonist administration and receiving at least medium-dose daily ICS (≥400 µg fluticasone or equivalent) with or without another controller, including chronic oral corticosteroids (OCS; prednisone ≤10 mg/day or equivalent) were enrolled. Individuals with known hypereosinophilic syndrome, a confounding underlying lung disorder or who had smoked within the last 6 months of screening were excluded.
In this post-hoc analysis, patients with RA were selected based on the definition outlined by the American Thoracic Society (ATS) workshop.6 Major inclusion criteria were OCS treatment (continuous or ≥50% of a 12-month period), and/or treatment with high-dose ICS (defined using the ATS/European Respiratory Society (ERS) guidelines on severe asthma).6 7 Additionally, patients had to fulfil at least two minor criteria: a controller medication in addition to ICS; persistent airflow obstruction (FEV1 <80% predicted) or ≥3 CAEs within the past 12 months.6 These three minor criteria were chosen from the seven in the ATS recommendations because there was insufficient historical data available to confirm the presence of others.6
Patient and public involvement
Patients were not involved in the design, conduct or interpretation of this post-hoc analysis.
Procedures
Each study consisted of a 2–4 week screening period and a 52-week treatment period, during which patients received either reslizumab 3 mg/kg or matching placebo as an intravenous infusion every 4 weeks.
Outcomes
The primary endpoint was the rate of CAEs, defined by: requirement for the use of SCS (oral, intramuscular or intravenous) in patients not already taking such treatment; a ≥2-fold increase in the dose of ICS or SCS for ≥3 days; or the need for asthma-related emergency treatment (emergency room visit, hospitalisation or unscheduled physician office visit for urgent treatment). Additionally, at least one of the following criteria must have been met: ≥20% decrease in FEV1; ≥30% decrease in peak expiratory flow rate on two consecutive days; or a worsening of symptoms or other clinical signs. Subanalysis determined the CAE rate defined by the requirement for asthma-related hospitalisation and/or use of SCS in patients not already receiving treatment or an increase from the baseline dose of SCS for ≥3 days.
Secondary endpoints included changes from baseline in FEV1; Asthma Quality of Life Questionnaire (AQLQ) score; ACQ score and Asthma Symptom Utility Index (ASUI) score. Safety was assessed by AEs (coded according to the Medical Dictionary for Regulatory Activities).
Statistical analyses
As this was a post-hoc analysis, it was not based on a pre-specified subgroup; no power calculations were conducted. All patients who received study drug were included in the intention-to-treat population and safety population. The CAE rate (events/patient/year) was based on a negative binomial (NB) regression model adjusted for stratification factors (baseline OCS use (yes or no) and geographical region (USA or other)). The ratio of CAE rate between the treatment groups and its 95% CI was estimated from the NB model. For the secondary efficacy endpoints, inferential statistics for mean changes from baseline over 52 weeks used a mixed model repeat measurement, with treatment, study, visit, treatment by visit interaction and stratification factors as fixed effects, and covariates for baseline value and patients as random effects. No formal statistical tests were planned for the safety analysis.
Results
Of the 953 patients randomised to receive either placebo or reslizumab 3 mg/kg in the two previous duplicate trials between 22 March 2011 and 9 April 2014, 306 (32%) met the criteria for RA (placebo, n=161; reslizumab, n=145).17 The Consolidated Standards of Reporting Trials diagram was presented previously.18 Baseline demographics, lung function and other characteristics were similar between groups (table 1).
Summary of baseline demographic and disease characteristics
The mean baseline dose of ICS (fluticasone-equivalent) received by patients with RA was 1080.8 µg, compared with 747.2 µg in the overall population (online supplementary figure 1A). Among patients with RA, 88% (of patients in both reslizumab and placebo groups) were receiving high-dose ICS, compared with only 43% and 44% of patients in the overall population, in the reslizumab and placebo groups, respectively. Similarly, 94% and 96% of patients with RA were taking a LABA in the placebo and reslizumab groups, respectively, but only 80% of those on placebo and 83% on reslizumab in the overall patient population. The mean baseline (prednisone-equivalent) dose of maintenance OCS was markedly higher among patients with RA (2.217 mg) compared with the overall population (0.851 mg) (online supplementary figure 1B). Of note, 30% of patients with RA received maintenance OCS, compared with 11% of patients in the overall population.
Supplemental material
Compared with placebo, patients with RA had a 59% (95% CI 40% to 72%) reduction in the adjusted mean rate of CAEs on reslizumab (figure 1A). The reduction in the overall population was 54% (42% to 63%).17 In the subanalysis of patients with RA, reslizumab resulted in a 59% (39% to 73%) reduction in the adjusted mean rate of CAEs requiring hospitalisation and/or use of SCS for ≥3 days compared with placebo (figure 1B). The corresponding reduction in the overall population was 57% (45% to 66%).

Adjusted mean rates of (A) CAEs and (B) CAEs requiring hospitalisation and/or use of systemic corticosteroids for ≥3 days over 52 weeks of treatment with placebo and reslizumab. Values in brackets below stated percentage differences represent 95% CI of percentage difference. CAEs, clinical asthma exacerbations.
Improvements in lung function (change from baseline in FEV1) were 158 mL (76–240) greater with reslizumab than placebo in patients with RA (figure 2). In the overall population, the difference was 110 mL (66–154).17
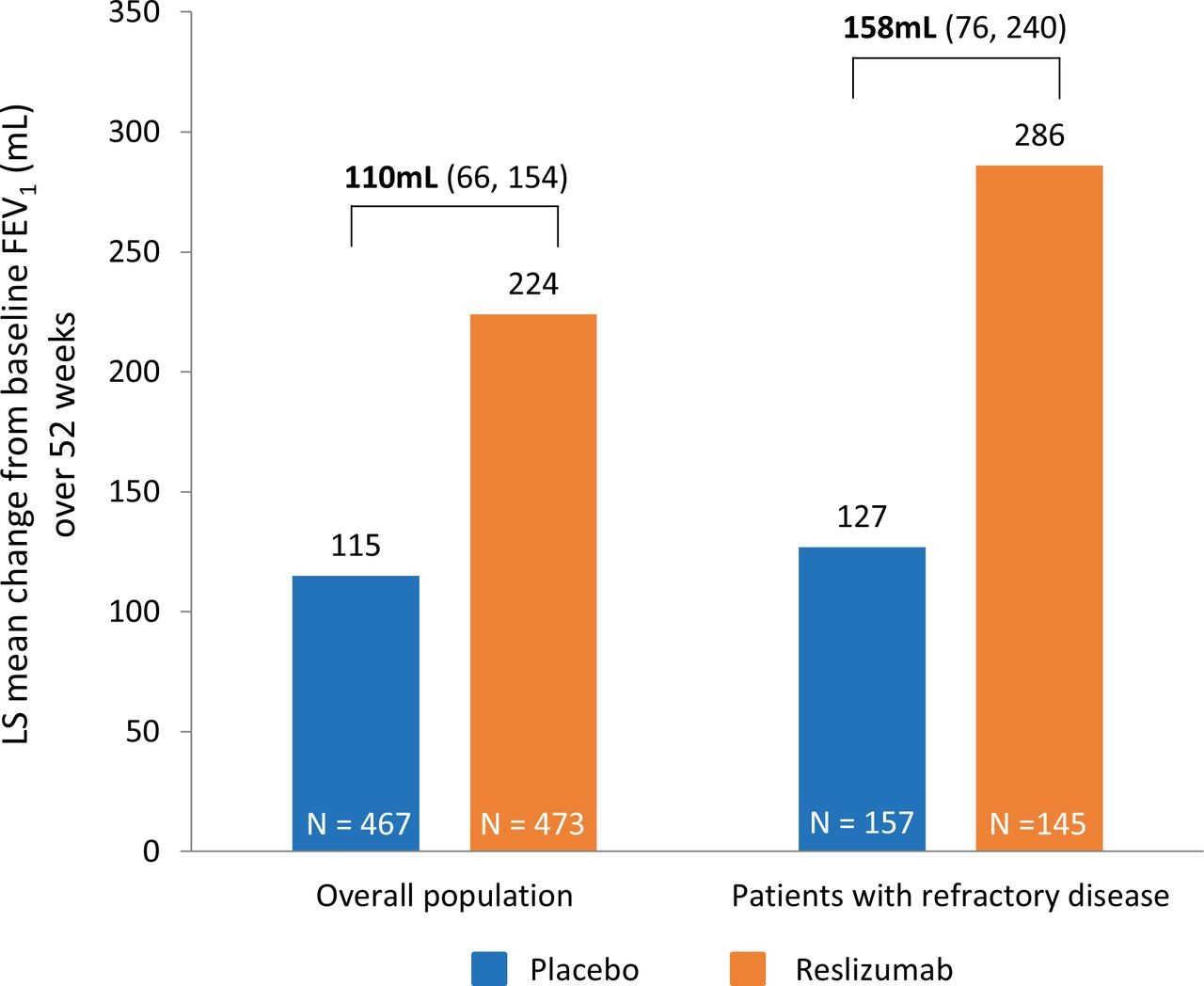
Lung function (FEV1): mean change from baseline over 52 weeks in patients treated with placebo or reslizumab. Values in brackets beneath treatment differences indicate 95% CI of treatment difference. CI, confidence interval; FEV1, forced expiratory volume in 1 s.
Compared with placebo, reslizumab improved patient-reported asthma control, asthma-related quality of life and asthma symptoms in the population with RA (figure 3). Changes from baseline in ACQ-7, AQLQ and ASUI scores were greater with reslizumab than placebo in the RA subgroup (figure 3) and the overall population.17

{kind=link}
{kind=link}
{kind=link}
Adjusted mean changes in (A) ACQ-7, (B) AQLQ and (C) ASUI scores over 52 weeks in placebo-treated or reslizumab-treated patients. Values in brackets beneath treatment differences in 95% CI of treatment difference. ACQ, Asthma Control Questionnaire; AQLQ, Asthma Quality of Life Questionnaire; ASUI, Asthma Symptom Utility Index; CI, confidence interval.
The observed safety profiles of reslizumab and placebo were similar (table 2). The overall rates of serious AEs, discontinuations due to AE and treatment-related AEs were similar in each treatment arm. The proportions of patients experiencing an AE of upper respiratory tract infection were slightly higher in the placebo group compared with the reslizumab group. However, this is of unclear clinical significance given that incidence rates for upper respiratory tract disorders (excluding infections) and upper respiratory tract signs and symptoms were similar with reslizumab and placebo. No specific AE displayed an incidence with reslizumab more than 2% above that seen with placebo. The pattern of frequency and severity of AEs in the RA subgroup was similar to that observed in the overall population.17
Incidence of adverse events. Data shown are numbers of patients (%)
Discussion
This post-hoc analysis provides the first evidence that reslizumab is effective and well tolerated in patients with more severe, treatment-RA and eosinophilia. Consistent with reported findings across all five phase 3 reslizumab studies,16–20 and in patient subgroups such as elderly patients,21 those with nasal polyps,22 late onset asthma23 and patients on maintenance OCS treatment,17 24 reslizumab significantly improved all efficacy parameters in patients with RA, including a reduction in asthma exacerbations, and improvements in lung function and patient-reported outcomes. Reslizumab was well tolerated overall and in the RA subgroup, displaying a safety profile similar to placebo in patients with RA despite their underlying severe refractory disease.
Although this is a post-hoc analysis, the clinical implications are very important as treatment options for patients with RA are limited. Long-term therapy with OCS has been the most common modality, but is associated with severe side effects.11 Biological therapies are being investigated as a possible means of addressing the treatment gap, including anti-IL-5 treatments such as reslizumab, mepolizumab and benralizumab. A small, single-centre, randomised, placebo-controlled trial of mepolizumab was performed in 61 patients with RA and eosinophilia.25 The study showed a reduction in the mean number of severe exacerbations per subject with mepolizumab treatment compared with placebo and improvement in AQLQ score, but no significant differences in lung function or symptom scores. However, the average number of peripheral blood eosinophils in this study was lower.25 Mepolizumab was subsequently investigated in the DREAM trial, which recruited 621 patients who met the ATS criteria for RA with severe asthma exacerbations and evidence of eosinophilic inflammation.6 26 Mepolizumab was effective in reducing clinically significant exacerbations compared with placebo, with reductions of 48% in the 75 mg group, 39% in the 250 mg group and 52% in the 750 mg group.26 Additional phase 3 trials have further evaluated mepolizumab in patients with severe asthma who met the ATS criteria for RA.6 27–29 In the MUSCA trial, mepolizumab was associated with clinically and statistically significant improvements in lung function at week 24 compared with placebo.29 The efficacy of benralizumab was investigated in the ZONDA trial, which included patients relying on OCS to manage severe asthma associated with eosinophilia, and who met the ATS criteria for RA.6 30 Benralizumab 30 mg reduced OCS use and exacerbation rates compared with placebo, although no significant effect on FEV1 was observed.30 Benralizumab was further investigated in the SIROCCO and CALIMA double-blind, placebo-controlled, phase 3 trials, which enrolled patients with severe asthma that was uncontrolled with high-dose ICS (≥500 µg/day fluticasone propionate or equivalent) plus LABA, and elevated eosinophils.31 32 Benralizumab, dosed subcutaneously every 4 weeks, significantly reduced annualised exacerbation rates over 48 weeks in the SIROCCO trial and over 56 weeks in CALIMA,31 32 and significantly improved prebronchodilator FEV1 compared with placebo in study weeks 48 and 56, respectively.31 32 Similar to the two double-blind, placebo-controlled, phase 3 trials from which the data for this analysis were obtained,17 although the inclusion criteria of SIROCCO and CALIMA did not fully meet the ATS criteria for RA,6 31 32 a subset of patients included in the trials who received high-dose ICS plus LABA, with or without OCS, and had persistent airway obstruction and asthma symptoms requiring short-acting beta agonist use are likely to have had RA. In the absence of head-to-head comparative studies of anti-IL-5 treatments in severe eosinophilic asthma, a number of recently published meta-analyses have indirectly compared the relative efficacy of reslizumab, mepolizumab and benralizumab.33–38 However, heterogeneity between the primary studies used in these meta-analyses, in terms of study design and patient eligibility criteria, particularly for whether patients had RA, means that it continues to be difficult to compare and rank the relative efficacies of anti-IL-5 treatments for asthma phenotypes.
The present study provides the first evidence that reslizumab may be an appropriate treatment for patients with severe RA and elevated blood eosinophil levels, resulting in improvements in exacerbations, lung function, asthma control and quality of life. It is noteworthy that the overall effects in the parameters investigated were not inferior (and, in fact, were numerically even higher) in the patient subgroup with RA compared with the overall population, suggesting that increased disease severity does not diminish treatment response to reslizumab. Indeed, it should not be assumed that a greater asthma severity is associated with greater efficacy in anti-IL-5 treatments. Previous evidence suggests that reslizumab may be more effective than subcutaneous mepolizumab in patients with severe, OCS-dependent asthma, and that higher doses of subcutaneous mepolizumab may be required to adequately clear airway eosinophils associated with OCS-dependent asthma.39 The similar findings between patient cohorts indicate that the value of reslizumab is not restricted to guideline-defined definitions of disease, but instead is determined by specific disease clinical phenotypes and biomarkers, such as eosinophilia and inadequate control on standard of care therapy. It is likely that the enrolment criterion of blood eosinophils ≥400 cells/µL in the overall population increased the probability that these patients had eosinophil-driven asthma and were, therefore, the most appropriate population for treatment with an anti-IL-5 antibody. Without this enrolment criterion, the treatment benefit of reslizumab observed in the OCS-dependent RA sub-population would likely have been higher than in the overall population, as OCS-dependent patients typically have higher EOS (eosinophil) levels.39 40 In the RA cohort, exclusion of emergency room visits, unscheduled physician office visits and the additional criteria for symptoms from the definition of CAEs had little impact on the rate ratio for reslizumab versus placebo. This suggests that the predominant driver of CAEs in this cohort is the requirement for hospitalisation and/or use of SCS, reflecting the severity of RA.
The main limitation of this study is that it was performed as a post-hoc subgroup analysis. Patients were classified as having RA based on the ATS definition6; however, definitions of high daily doses of ICS were obtained from the international ATS/ERS guidelines of severe asthma, which reflect more recent formulations. The availability of historical data for only three of the seven minor criteria that comprise the ATS workshop definition of RA restricted the number of potential patients that could be included in the analysis. The original studies were not powered to assess reslizumab in patients meeting the criteria for RA, and it would, therefore, be of interest to conduct a prospective, randomised, controlled trial specifically in this population. It would also be valuable to include a longer follow-up period than 52 weeks to assess long-term efficacy and safety. Strengths of this study include the substantial number of patients with RA, the randomised, double-blind, placebo-controlled study design of the parent studies, and the inclusion of a range of well-established, clinically- and patient-relevant efficacy endpoints.
In conclusion, the current analysis suggests that add-on therapy with intravenous reslizumab 3.0 mg/kg provides clinical benefit in patients with RA and eosinophilia and is well tolerated. Significant improvements in asthma outcome measures that were numerically greater than those seen in the overall population, were observed in this sub-population despite higher baseline ICS doses and a higher proportion of OCS-dependent patients at baseline compared with the overall population. Our finding of significant benefit across all efficacy measures in this severe subgroup of patients with RA is consistent with prior studies of reslizumab in patients with asthma and eosinophilia.
References
Footnotes
Contributors All authors of this manuscript were involved in the acquisition, analysis or interpretation of the data, and in the preparation or critical revision of the manuscript. All authors approved the final version of the manuscript.
Funding This study was sponsored by Teva Branded Pharmaceutical Products R&D, Inc. Medical writing support was provided by Ian C. Grieve, PhD, of Zoetic Science (an Ashfield company, part of UDG Healthcare plc), and was funded by Teva Branded Pharmaceutical Products R&D, Inc. (West Chester, Pennsylvania, USA) during the preparation of this manuscript.
Competing interests JCV has lectured for and received honoraria from Allergopharma, ALK, Asche/Chiesi, AstraZeneca, Avontec, Bayer, Bencard, Bionorica, Boehringer Ingelheim, Cipla, Essex/Schering-Plough, GSK, Janssen-Cilag, Laboratorios Leti, Meda Pharmaceuticals, Merck/MSD, Mundipharma, Novartis, Nycomed/ALTANA, Pfizer, Regeneron, Revotar Biopharmaceuticals, Sandoz/Hexal, Sanofi, Stallergenes, Takeda, Teva, UCB/Schwarz Pharma, Zydus Cadila and possibly others and has participated in advisory boards for Asche Chiesi, Avontec, Boehringer Ingelheim, Essex/Schering-Plough, GSK, Janssen-Cilag, MSD, Mundipharma, Novartis, the Paul-Ehrlich-Institute, Revotar Biopharmaceuticals, Sandoz/Hexal, Sanofi, Takeda, Teva, UCB/Schwarz Pharma and possibly others and has received grants from the Deutsche Forschungsgemeinschaft, Land Mecklenburg-Vorpommern, GSK., and MSD. MM was an employee of Teva Pharmaceuticals at the time the analyses were conducted. MG is an employee of Teva Pharmaceuticals. SK has received consulting and lecture fees from Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, Teva and Roche.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval Both studies were done in accordance with Good Clinical Practice guidelines, the Declaration of Helsinki and local regulatory requirements. Relevant health authorities and local ethics committees or institutional review boards approved the study protocols and patients provided written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The data sets used and/or analysed for the study described in this manuscript are available upon reasonable request. Qualified researchers may request access to patient-level data and related study documents including the study protocol and the statistical analysis plan. Requests will be reviewed for scientific merit, product approval status and conflicts of interest. Patient-level data will be de-identified and study documents will be redacted to protect the privacy of trial participants and to protect commercially confidential information. Please email USMedInfo@tevapharm.com to make your request.