Article Text

Abstract
Background Patients with idiopathic pulmonary fibrosis (IPF) have significantly higher incidence of lung cancer (LC) relative to the general population. There is a further increase in LC incidence in patients with IPF subsequent to lung transplant, specifically in patients with IPF undergoing single lung transplant.
Objectives To examine the incidence and characteristics of LC in patients with IPF during follow-up and after lung transplantation (LTX).
Methods We conducted a retrospective analysis of all patients with IPF diagnosed with LC in Rabin Medical Center, Israel, over an 11-year period. We compared the characteristics of transplanted patients with IPF diagnosed with LC to patients with IPF who did not undergo lung transplant. Data were accessed from database registries using the words ‘fibrosis’, ‘lung-cancer’ and ‘lung-transplantation’. Demographic parameters included age, gender and smoking history (pack years). Clinical-pathological parameters included lapse in time from IPF diagnosis to LC, type of malignancy, affected pulmonary lobe, and stage at diagnosis, oncological treatment and survival.
Results Between 2008 and 2018, 205 patients with IPF underwent lung transplantation at our medical centre. Double LTX was performed in 83 and single LTX in 122 cases. Subsequently, 15 (12.3%) single LTX patients were diagnosed with LC during the study period. During the same period, of 497 non-transplanted patients with IPF followed in our centre, 45 (9.1%) were diagnosed with LC. In all 15 transplanted patients with IPF, LC was diagnosed exclusively in the native fibrotic lung. LC incidence was higher in the transplanted as compared with the non-transplanted group, but this difference did not reach statistical significance (OR=0.7, 95% CI 0.38 to 1.32, p=0.28). At LC diagnosis, the non-transplanted group was older than the transplanted group with average age of 67.7 versus 60.8 years, respectively (p=0.006). Both groups showed male predominance. In both groups, LC was primarily peripheral, lower lobe predominant and most frequently squamous cell carcinoma. The median survival time after LC diagnosis was 4 months in the transplanted group and 11 months in the non-transplanted group (p=0.19). Multivariate analysis showed improved survival in the non-transplanted group among those patients who received oncological treatment.
Conclusion Chest CT should be performed regularly in order to evaluate IPF patients for potential LC. Single lung transplant IPF patients face an increased risk of post-transplant LC in the native fibrotic lung. Where practicable, IPF patients should be prioritised for double lung transplant.
- interstitial fibrosis
- lung cancer
- lung transplantation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
This was a single center retrospective observational study of transplanted and non-transplanted IPF patients over a decade of follow up in a tertiary medical center
This was a retrospecive observational study of more than a decade follow up with transplanted and non transplanted IPF patients in a single tertiary center
Lung cancer in patient with IPF has unique characteristics, with the cancer being primarily peripheral, lower lobe predominant and most frequently squamous cell carcinoma.
The reader will find in our study that the population of transplanted and non-transplanted patients with IPF have different clinical, histological and prognostic characteristics making this a unique population.
Background
Idiopathic pulmonary fibrosis (IPF) is a known risk factor associated with increased incidence of lung cancer (LC).1–3 While LC prevalence in the general population is estimated at 0.007%–0.016%, epidemiological studies in patients with IPF have demonstrated LC incidence to range from 4.4% to 13.0%.4 5 While the mechanism of transition from fibrotic epithelial cell to malignant cell is not entirely clear, it is assumed to be associated with genetic and epigenetic changes and altered cell-to-cell communication, which stimulate uncontrolled cell proliferation and the formation of malignant cells.6–9
Patients with IPF have a poor prognosis, with an average expected survival of 5–6 years from diagnosis.10 11 For many patients with end-stage IPF patients, lung transplant remains the only option for increasing life expectancy.12 13 However, the immunosuppressive regimen required post-transplant increases the risk for LC,14 15 which is even more pronounced in single lung transplant recipients, as the remaining native fibrotic lung can be a nidus for de novo malignancy.16
Over the last decade, we have observed in our medical centre an increase in the incidence of LC in patients with IPF after transplant as compared with non-transplanted IPF patients. We conducted this study to assess the incidence and characteristics of LC in transplanted as compared with non-transplanted IPF patients.
Methods
Study design and definitions
We conducted a retrospective analysis of all patients with IPF diagnosed with LC in Rabin Medical Center, Israel, from January 2008 to December 2018. IPF cases were diagnosed by biopsy (surgical resection, transbronchial forceps and cryoprobe) with pathological reading of usual interstitial pneumonia pattern or radiological pattern meeting criteria for IPF based on the then current guidelines at the time of diagnosis.
In our medical centre, prior to the 2011 consensus guidelines,17 IPF was diagnosed by the treating pulmonologist based on a constellation of clinical, radiological and histological constellation findings. After 2011, IPF diagnosis was made by a multidisciplinary board consensus based on published guidelines.
Before 2011 guidelines,17 IPF was diagnosed based on clinical, radiological and histological constellation at the discretion of the providing pulmonologists. After 2011 IPF diagnosis was made by a multidisciplinary board consensus. Diagnosis of LC was made based on pathological findings from biopsies performed using bronchoscopy (endobronchial or transbronchial biopsies), surgical resection, pleural fluid analysis or imaging guided needle biopsies. All cases of LC were presented and discussed at a multidisciplinary tumour board session for decision on management. Survival was calculated from the date of cancer diagnosis to the date of death.
Standard follow-up
In our medical centre, non-transplanted IPF patients underwent chest CT for LC screening at intervals determined at the discretion of the treating physician. Transplanted patients were followed by transplant specialists with a comprehensive protocol that included clinic visits, chest imaging, blood tests, pulmonary function testing and lung allograft biopsies. Transplant patients were seen in clinic weekly for the first month post-transplant, then twice a month for 3 months and subsequently once every 2 months. At each clinic encounter, patients performed spirometry and routine blood tests, including immunosuppressive medication level, complete blood count and basic metabolic panel. Chest X-ray were performed daily postoperatively until discharge, followed by chest CT and lung perfusion scans every 6 months for the first year post-transplant and once annually thereafter. Bronchoscopy with allograft biopsies were performed to detect rejection at 72 hours postoperatively (inspection only), 2 weeks, 1 month and 3 months and thereafter depending on clinical need. All LC cases were also followed by the thoracic oncology unit with case-by-case clinic visits, chest imaging and blood tests.
Data collection and analysis
Patient data were obtained from our medical centre database registries, including searches using the words ‘fibrosis’, ‘IPF’, ‘lung-cancer’ and ‘lung-transplantation’ (database sources Ofek, Rabin Medical Center-PACS, Chamelion, Microsoft Access lung transplant database and OncoPro). Demographic parameters included age, gender and smoking history (pack years). Clinical-pathological parameters included time elapsed from IPF diagnosis to LC diagnosis, type of malignancy, affected pulmonary lobe, cancer stage at diagnosis, oncological treatment and survival.
Statistical analysis
Statistical analysis for categorical variables was performed by using χ2 test, while continuous variables were compared using t-test. The value of p<0.05 was considered statistically significant. Survival analysis was presented as univariate analysis with HRs, and multivariate analysis was performed and presented including all variables with potential to impact survival of transplanted and non-transplanted patients. Statistical analysis was performed using SPSS V.18.0 software.
Patient and public involvement
This research was done without patient involvement. Patients were not invited to comment on the study design and were not consulted to develop patient relevant outcomes or interpret the results. Patients were not invited to contribute to the writing or editing of this document for readability or accuracy.
Results
Demographic
From January 2008 to December 2018, 15 transplanted IPF patients and 45 non-transplanted IPF patients at our medical centre were diagnosed with de novo LC. The clinical and demographic characteristics of those patients are presented in table 1.
Clinical and demographic characteristics
During the study period, our center followed 205 patients with IPF who underwent lung transplant. Of these, 122 received a single lung transplant and 83 received a double lung transplant. Out of the single lung transplants, 15 (12.3%) patients were diagnosed with LC during follow-up, whereas no cases of LC were diagnosed in the double lung transplanted group. All cases of LC in the transplant group were diagnosed in the native lung. During the same time period, 497 non-transplanted IPF patients were followed in our center, of them 45 (9.1%) were diagnosed with LC (figure 1). Though the incidence of LC in the transplanted IPF group was higher than in the non-transplanted IPF group, this difference did not reach statistical significance (OR=0.7, 95% CI 0.38 to 1.32, p=0.28).
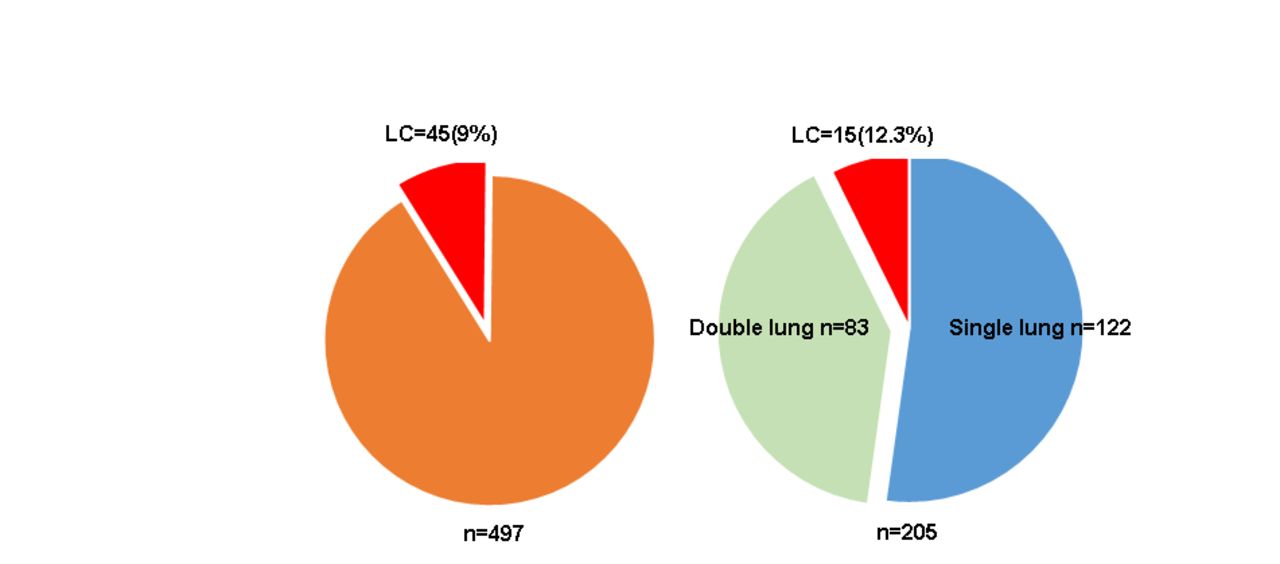
Lung cancer (LC) diagnosis in non-transplanted IPF patients (left) versus transplanted IPF patients (right). LC cases presented as red coloured slices.
Non-transplanted IPF patients were older on average than the transplanted IPF patients (mean 67.7 vs to 60.8, p=0.006). There was a male predominance with 87.5% (40) in the non-transplanted IPF group and 66.7%10 in the transplanted IPF group. The average smoking history (measured in pack years) was higher in the non-transplanted IPF group than in the transplanted IPF group (35 compared with 20, p=0.035).
Lung cancer characteristics
Notably, all 15 IPF transplant patients with LC had undergone single lung transplantation, and the LC was diagnosed in the native fibrotic lung; none presented with LC in the allograft lung.
LC tumours were more commonly located in the lower lobes of either lung with 62.2% (28 cases) and 53.3% (8 cases) in the non-transplanted groups and transplanted group, respectively (p=0.66). Peripheral location of LC was more common than central in both the non-transplanted IPF group, 73.3% (33 cases), and in the transplanted IPF group 66.7% (10 cases) (p=0.42). Table 2 presents the tissue diagnosis modalities used for LC diagnosis in our cohort. Transthoracic needle biopsy was the most common method used in both groups, with 62.2% in the non-transplanted IPF group and 33.3% in the transplanted IPF group. In the transplanted IPF group, a third of LC cases presented as pleural effusion and diagnosis was made by thoracentesis and fluid analysis.
Lung cancer tissue diagnosis modalities
Squamous cell carcinoma (SCC) was found in 42.2% of non-transplanted patients, followed by 37.8% with adenocarcinoma (AC), the difference was not statistically significant (p=0.9). A similar trend for SCC and AC was found in the transplanted group (40% vs 26.7%, respectively). Small cell carcinoma was detected in two patients in the transplanted group (13.3%) and four in the non-transplanted group (8.9%). One case of large cell carcinoma was diagnosed in the non-transplanted group.
In the non-transplanted group, six patients (13.3%) were diagnosed simultaneously with IPF and LC. All six patients were included in the non-transplanted group for this analysis. They had no significant difference in treatment and disease progression compared with other patients in the non-transplanted IPF group.
The average time interval from IPF diagnosis to LC diagnosis in the non-transplanted IPF group was 3 years, significantly shorter than 10 years in the transplanted IPF group (p<0.001).
LC was most frequently diagnosed at an advanced stage (III–IV), with 66.7% of transplanted IPF patients diagnosed in stage IV, as compared with 44.4% of non-transplanted IPF patients.
Treatment and survival
Of the transplanted group, 66.7% (10 patients) received oncological treatment as compared with 75.6% (34 patients) in the non-transplanted group (p=0.18).
LC therapy modalities were similar for both groups, with the exception of immunotherapy, which was not offered to transplanted IPF patients (table 3). One-third of the transplanted IPF group did not receive oncology therapy at all, as compared with 24.4% in the non-transplanted IPF group.
Lung cancer treatment
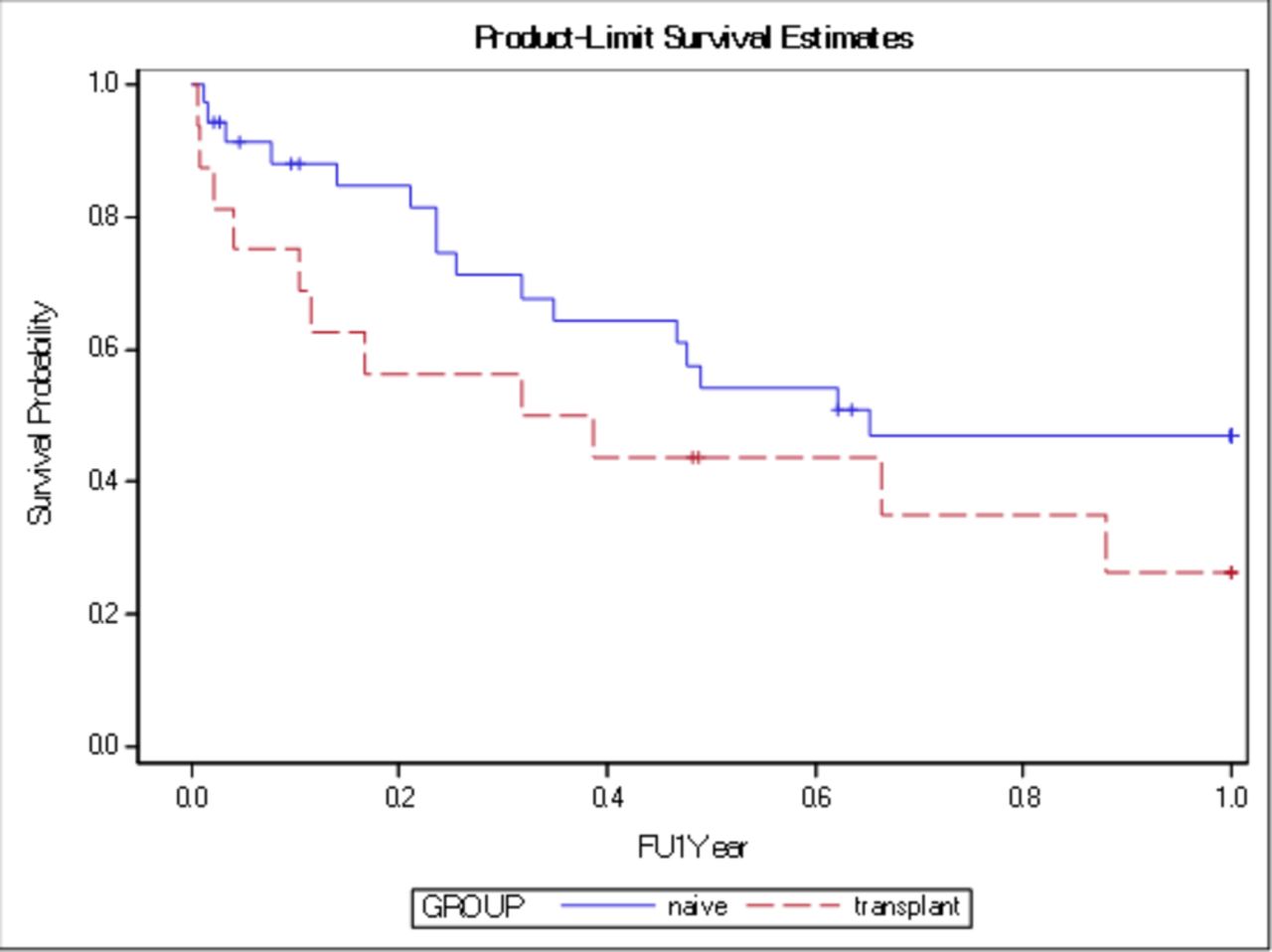
Median survival after LC diagnosis was 4 months in the transplanted IPF group, as compared with 11 months in the non-transplanted IPF group (p=0.19). Survival exceeded 12 months for 27% of the transplanted IPF group, as compared with 43% of the non-transplanted IPF group (p=0.26) (figures 2 and 3).
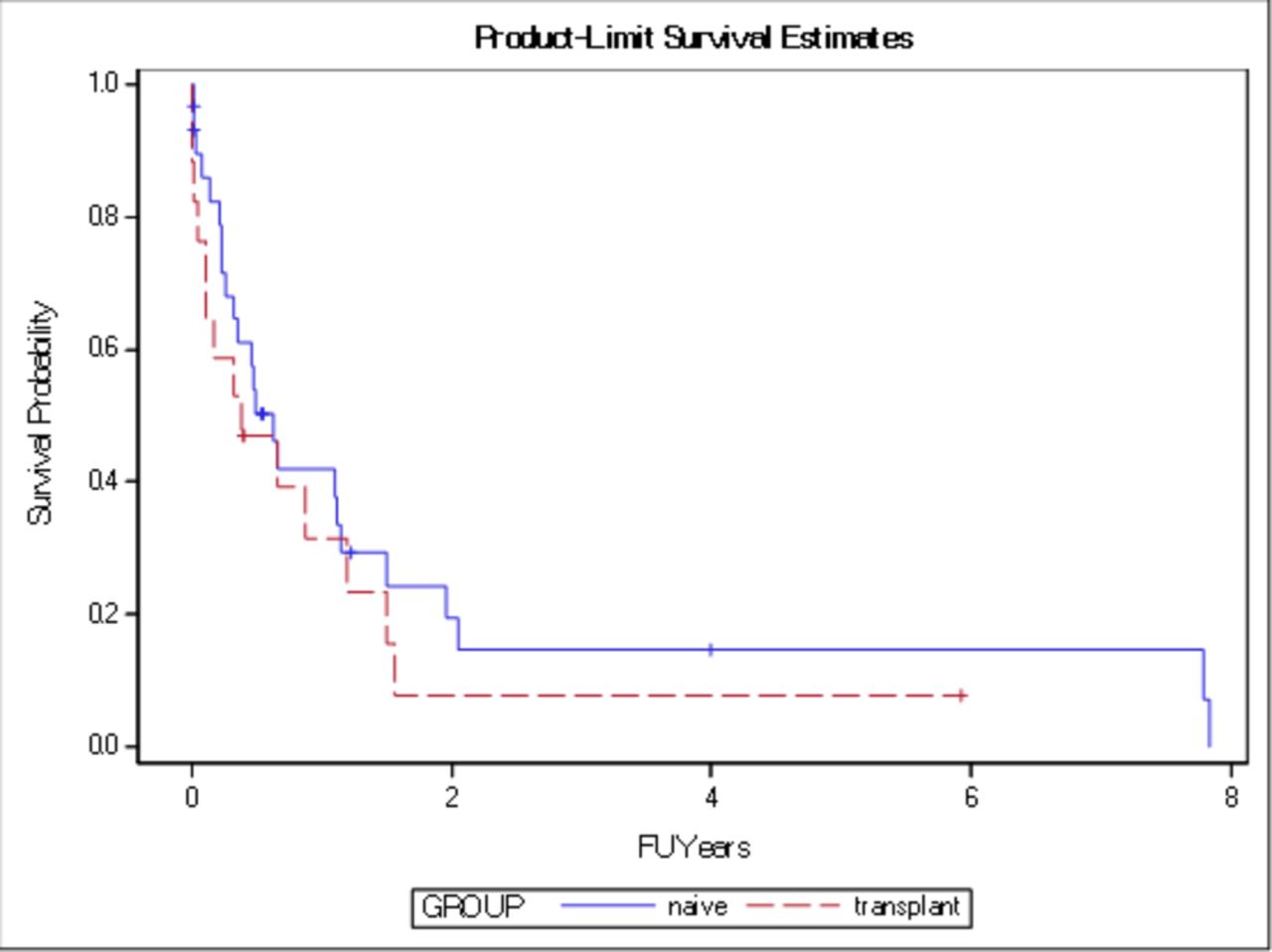
Kaplan-Meier general survival curve for both groups transplanted and non-transplanted (naïve) without significant difference in median survival (p=0.19).FU=follow up
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier 1-year survival curve for both groups transplanted and non-transplanted (naïve) without significant difference (p=0.26). FU=follow up
In the non-transplanted IPF group, six patients were diagnosed with LC in an explant lung removed during lung transplantation. Excluding one patient who died at the time of transplantation, two were alive at the end of the study, and the three remaining had an average survival of more than 2 years, with none having LC recurrence.
Four non-transplanted IPF patients were receiving treatment at the time of LC diagnosis with Nintedanib (Boehringer Ingelheim), a tyrosine kinase inhibitor with antifibrotic and antineoplastic activity. None of the patients in our cohort received any other antifibrotic medication.
Multivariate analysis, adjusted for age, gender and smoking exposure, demonstrated that advanced stage (III–IV) at diagnosis and squamous cell histology were associated with shorter survival in the non-transplanted IPF group with HR=5.49 (95% CI 2.025 to 14.908) and HR=5.54 (95% CI 1.24 to 24.44), respectively. Moreover, survival advantage was demonstrated only for the non-transplanted IPF group receiving oncology treatment (HR=0.215, 95% CI 0.079 to 0.584).
Discussion
In this study, we have presented our experience with IPF patients diagnosed with LC, both during routine medical follow-up as well as postlung transplantation. To our knowledge, this series is the largest cohort of transplanted IPF patients with LC.
Demographic data in this cohort, including gender distribution, age at LC diagnosis and smoking history, are similar to demographics described in a prior study of patients with IPF.18 Similarly, average time interval from IPF diagnosis to LC diagnosis in prior studies19–21 was 30–52 months for non-transplanted IPF patients, as compared with 36 months for non-transplanted IPF patients in our cohort. In contrast, in our cohort, the median time interval from IPF diagnosis to LC diagnosis in the transplanted IPF patients was 10 years. This may relate in part to the relatively younger average age of the transplanted IPF patients, such that the difference in time interval to LC diagnosis is due to their younger age, rather than their lung transplant. In addition, in our cohort, the non-transplanted IPF group had a statistically significant longer smoking history as compared with the transplanted IPF group.
LC in patients with IPF more frequently develops in lung regions affected by fibrosis, with a consequential preference in patients with IPF for peripheral regions.22–25 Indeed, our cohort demonstrated a peripheral predominance, including 3 of 4 diagnoses of small cell carcinoma in the lung periphery.
We found that squamous cell carcinoma was the most frequent histology, followed by adenocarcinoma, in contrast to the predominance of adenocarcinoma histology in LC diagnosed in the general population.26 The greater incidence of squamous cell carcinoma in patients with IPF may relate to similarities in the pathogenesis of transition from reactive inflammation to fibrosis and subsequently to squamous cell carcinoma.
LC in IPF patients is often discovered at advanced stages (III–IV) and consequently has a poor prognosis. In one study, 61% of IPF patients diagnosed with LC were stage III–IV at diagnosis.21 Similarly, in our study population, 66.6% of non-transplanted IPF patients presented in advanced stages III–IV. Notably, almost three-quarter (73.4%) of the transplant IPF patients were diagnosed at stage III–IV, despite our protocol for annual chest CT screening for transplanted IPF patients. This relatively late stage LC detection in transplanted IPF patients implies that radiological manifestations of LC may be more challenging to detect in the native fibrotic lung, and immunosuppressive treatment may accelerate the progression of clinically unnoticed disease despite rigorous follow-up protocol.
Previous studies assessed whether earlier discovery of LC in patients with IPF can provide a survival benefit. Ozawa showed no difference in 5-year and 10-year survival for IPF patients with a new LC diagnosis who received oncological treatment compared with those not treated for LC.19 Furthermore, Khan et al found no survival benefit following oncology therapy for LC in patients with or without concomitant IPF.24 Interestingly, Tomassetti et al found lack of evidence for its benefit and raised concern regarding the potential risk of cancer treatment in these patients, due to an increased incidence of respiratory complications, specifically IPF exacerbation.21 In our study, multivariate survival analysis showed that for the non-transplanted IPF patients with LC, oncology therapy prolongs survival (p=0.003). Given the multiplicity of oncological treatments and the small sample size, we could not assess which oncology treatment could be beneficial and which may be hazardous. We suggest that the decision and choice of modalities to treat LC in IPF patients should be addressed on a case-by-case basis by a multidisciplinary team.
Yoon et al showed that patients with IPF have an increased incidence for LC among single allograft recipients (13 out of 97, 13.4%), with increased mortality compared with single allograft recipients without LC (p=0.028).18 Survival rates in our cohort were similar, with a median of 4 months in the transplanted IPF group as compared with 11 months in the non-transplanted IPF group. Our key finding was the development of LC only in IPF patients with single lung transplant; no double lung transplant recipients from our cohort developed LC during the 11-year study period. Moreover, none of the six patients with LC diagnosed in their explanted lung experienced a recurrence postlung transplantation. This suggests that lung transplantation may be still appropriate for patients with end-stage IPF disease despite a concurrent diagnosis of early stage LC. Moreover, double lung transplantation provides a de facto advantage in prevention of LC in patients with IPF through removal of the fibrotic lungs.
This single-centre study has several limitations. First, being retrospective in design, the study relied on the accuracy and fidelity of historical data recorded in our medical centre registries. Second, our medical center has no recommended protocol for follow-up of non-transplanted IPF patients, and chronicity of imaging for cancer screening is left to the discretion of the attending clinician. In contrast, the transplanted IPF group was closely monitored, including annual CT chest imaging at a minimum. Third, the diagnosis of IPF was based on the current consensus guidelines at the time of diagnosis; in retrospect, some of the patients with IPF in the study might have not fulfilled the current diagnostic criteria for IPF. Fourth, although the study included all IPF patients in our medical center over a decade, it was underpowered to detect significant difference between groups in most study parameters.
Our study showed that single lung transplant IPF patients are at increased risk for LC, which will more likely be diagnosed at an advanced stage with a worse prognosis. Patients with IPF considered for transplant should therefore be prioritised for double lung transplants. For IPF patients with single lung transplant, vigilant post-transplant screening should be implemented for early LC detection.
Acknowledgments
The authors would like to thank Mrs Dalia D Orkin for her important English language contributions and editing services.
References
Footnotes
Contributors All named authors had a substantial contribution to the conception and design of the work, the acquisition, analysis and interpretation of data. All authors are in approval of the version published. All authors are in agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. EG: principal investigator and responsible for study design and conception, data collection, results interpretation, statistical analysis, drafting, writing and submitting the manuscript. AZ: design and conception, data collection, oncological perspective, results interpretation, drafting and writing. BP: design and conception, data collection, results interpretation, statistical analysis, drafting and writing. MH, OS, DR and DS: design and conception, data collection, results interpretation, drafting and writing. MK: supervisor physician of the study and was responsible for designing the study and conception, results interpretation, drafting the article and revising its content.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information.