Article Text

Abstract
Introduction Airway clearance techniques (ACTs) are a gold standard of cystic fibrosis management; however, the majority of research evidence for their efficacy is of low standard; often attributed to the lack of sensitivity from outcome measures (OMs) used historically. This randomised controlled trial (RCT) investigates these standard OMs (sputum weight, forced expiratory volume in 1 s) and new OMs (electrical impedance tomography (EIT), multiple breath washout (MBW) and impulse oscillometry (IOS)) to determine the most useful measures of ACT.
Methods and analysis This is a single-centre RCT with crossover design. Participants perform MBW, IOS and spirometry, and then are randomised to either rest or supervised ACT lasting 30–60 min. MBW, IOS and spirometry are repeated immediately afterwards. EIT and sputum are collected during rest/ACT. On a separate day, the OMs are performed with the other intervention. Primary endpoint is difference in change in OMs before and after ACT/rest. Sample size was calculated with 80% power and significance of 5% for each OM (target n=64).
Ethics and dissemination Ethics approval was gained from the London–Chelsea Research Ethics Committee (reference 16/LO/0995, project ID 154635). Dissemination will involve scientific conference presentation and publication in a peer-reviewed journal.
Trial registration numbers ISRCTN11220163 and NCT02721498.
- cystic fibrosis
- respiratory measurement
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Over 10 500 people in the UK have cystic fibrosis (CF),1 a genetic disorder of ion transport across cell membranes causing organ damage with a median predicted survival of 47.3 years. Impaired mucociliary clearance in the CF lung due to airway dehydration and viscous secretions causes airway obstruction, mucus plugging and infections.2 Removal of airway secretions is critical to minimise recurrent infections and inflammation, which lead to lung damage, respiratory failure and death.3 4 Traditionally, combinations of mucoactive agents and airway clearance techniques (ACTs) are prescribed on an individual basis by specialised physiotherapists.5
There are many ACTs including breathing methods such as the active cycle of breathing techniques (ACBTs) or autogenic drainage and adjuncts such as positive expiratory pressure, high frequency chest wall oscillation or oscillatory devices, for example, the Acapella. Previous research comparing several ACTs showed no significant difference in clinical effectiveness between techniques over 1 year,6 and the Association of Chartered Physiotherapists in Cystic Fibrosis standards of care does not advocate one technique over the others based on the available evidence.7 However, the currently available research regarding ACTs is mainly of low quality on the GRADE (Grading of Recommendations, Assessment, Development and Evaluations) system used in systematic reviews,7 often due to methodological or outcome measurement issues.
A long-standing debate exists within respiratory physiotherapy and the wider medical community about which are the best outcome measures (OMs) for research into ACTs in CF.8 Established measures of forced expiratory volume in 1 s (FEV1) and expectorated sputum wet weight have been recognised by the international CF community as the gold standard for ACT research, although their limitations are well recognised.6–12
Spirometry, yielding the FEV1 is effort dependent, and is criticised as having high variability due to patient participation in the manoeuvre.11 The nature of completing a forced expiratory manoeuvre, such as FEV1, may create an airway reaction or mobilise sputum, which on its own, may influence ACT study results. FEV1 also mainly reflects obstruction of large airways, and can remain ‘normal’ when significant small airway damage is present.13 As the small airways are affected early in CF lung disease, OMs need to effectively monitor this area.14 In addition, studies using FEV1 may not detect differences as it may not be sensitive enough to pick up small, but possibly clinically significant changes.6–8 12
Weight of sputum expectorated is often used as a simple OM to reflect the amount of mucus mobilised during ACT.7 15 However, sputum expectorated may not be the most relevant or valid OM: it is not specific to airway clearance or alveolar recruitment; it is not sensitive to small changes or suitable in early disease; and it has limited repeatability, being dependent on the will/ability of a subject to fully expectorate. Sputum could be swallowed, cleared after ACT, or contaminated by saliva, all which can result in inaccurate estimations of ACT effect.7 8 16
Recent technological advances have supported the development of new OMs. These may provide more detailed information on ACT effect in relation to small airways function, ventilation distribution and changes in airway obstruction and include multiple breath washout (MBW), electrical impedance tomography (EIT) and impulse oscillometry (IOS).
The lung clearance index (LCI2.5) is calculated from an MBW, which measures the number of lung volume turnovers required to clear an inert tracer gas during relaxed tidal breathing. When the level of the tracer gas has fallen to 1/40th of the starting concentration, the washout is considered complete. Functional residual capacity (FRC) and the cumulative expired volume (CEV) can be derived from MBW measurements, and are used to calculate LCI2.5 (CEV divided by FRC).17 LCI2.5 reflects ventilation inhomogeneity, increasing with airway obstruction typical in more severe disease.18 LCI2.5 detects small changes in CF lungs, in particular in peripheral airways, and so is considered a sensitive measure of lung disease severity.18–20 Despite research into reliability, validity and responsiveness of LCI2.5 as a longer term OM, and support of LCI for clinical trial use by the European CF Society Clinical Trial Network Standardisation Committee,14 it is not clear whether LCI2.5 is useful in determining differences over a short time, for example, single-ACT sessions.14 21 22 In particular, as LCI2.5 is altered by airway obstruction, the movement of secretions which occurs during ACT, alongside altered ventilation of lung segments, could result either in increased or decreased LCI2.5; both directions of change have been reported in previous small studies measuring ACT effect.22 23
IOS provides information about the mechanical properties of airways by using alternating frequency pressure oscillations, in the form of sound waves, superimposed over normal breathing. Airflow and sound wave response are used to calculate resistance (R) and reactance (X) to tidal breathing.24 Changes in IOS, demonstrating changes in resistance to airflow indicate obstructions to the airways, for example, mucus plugging or altered airway reactivity such as in asthmatic airways.25 Low frequency oscillations, such as those at 5 Hz (R5), travel through the airways to the peripheries and are reflected back, while higher frequencies such as those at 20 Hz (R20) are reflected back from central airways.26 Calculation of the difference between R5 and R20 (R5−R20) indicates extent of small airway obstruction. Reactance (X) indicates the elastic recoil of the airways. Most literature is in other patient populations, such as asthma; however, there are some studies emerging using IOS as an endpoint for CF intervention evaluation.27 28
EIT creates a real-time image of ventilation by measuring the impedance to a low voltage electrical current as it travels through the lungs. Electrodes are attached to the thorax via a belt, and small alternating currents (of a magnitude undetectable to the subject) are applied. The resulting electrical potentials are measured, and a reconstruction algorithm is used to obtain the electrical impedance distribution within the thorax.29 Analysis of the data obtained calculates end expiratory lung impedance, which is affected by changes in lung volumes and ventilation, and so has been proposed as a measure to detect changes resulting from sputum mobilisation and ACT.6 30 Most current literature in EIT is from the intensive care environment,29 31 with few studies investigating EIT in CF.32–34 Wettstein et al34 demonstrated that EIT detected differences in ventilation distribution following ACT in a small cohort of nine CF adults, but reported high variability. These findings support further investigation of a larger sample of patients during ACT with a wide range of lung disease severity to assess whether the high intersubject variability observed is due to differences within the individual participants (eg, airway obstruction), or due to the inherent variability of EIT itself.
The importance of patient preference and patient reported OMs (PROMs) is acknowledged within CF ACT research.8 9 12 The most sensitive OM scientifically, may be the least useful clinically if patients do not wish to perform it. The present study, therefore, includes a PROM in the form of a questionnaire asking opinions on the OMs performed. Good patient feedback of an OM is the key to its future within CF research and clinical care.
Several Cochrane reviews suggest further work is required to identify the most appropriate OMs for ACT assessment.8 15 35–40 The European CF Society Allied Health and Nursing Professions Working Group reported the need for evaluation and identification of appropriate and robust OMs for allied health professional research as one of their four key objectives.41
This study seeks to identify a robust OM in ACT research that will benefit patients, facilitating their choice of ACT. Additionally, identifying the best OM will benefit the physiotherapy community in designing future trials of ACTs. Furthermore, industry can use this research in the assessment of new devices and mucoactive agents.
Our hypothesis is that the use of a new OM tool box will allow more sensitive assessment of ACTs in stable adult patients with CF than the current gold standard measures. It was developed following extensive literature review and discussions with CF experts and patients. These experts acknowledge that physiotherapy practice guidelines are largely based on low-grade evidence because of underpowered studies with poor OMs.7 42 This study seeks to improve this situation by evaluating new OMs thoroughly, providing robust evidence, which could then be used in future studies.
Methods and analysis
Trial registration
This trial is registered on the public database www.clinicaltrials.gov.uk and is part of the International Standard Randomised Controlled Trial Number (ISRCTN) database.
Patient involvement
Patients were involved in the conceptualisation, design and conduct of this research. During planning patients’ opinions on the trial design, research question and recruitment methods were gathered via discussions with adults with CF, and the Patient Representatives Group (RPG) of the Respiratory Clinical Research Facility (RCRF) based at the Royal Brompton Hospital (RBH), London. The study assessment questionnaires (online supplemental appendices 1 and 2) have been designed and developed in conjunction with the CF patient advocate at RBH and the RPG. A patient representative is an independent member of the trial steering committee.
Supplemental material
Supplemental material
Study design
This is an open label, single-centre, single-blind, randomised controlled crossover trial investigating OMs for ACT in stable adults with CF. Figure 1 illustrates the study design.
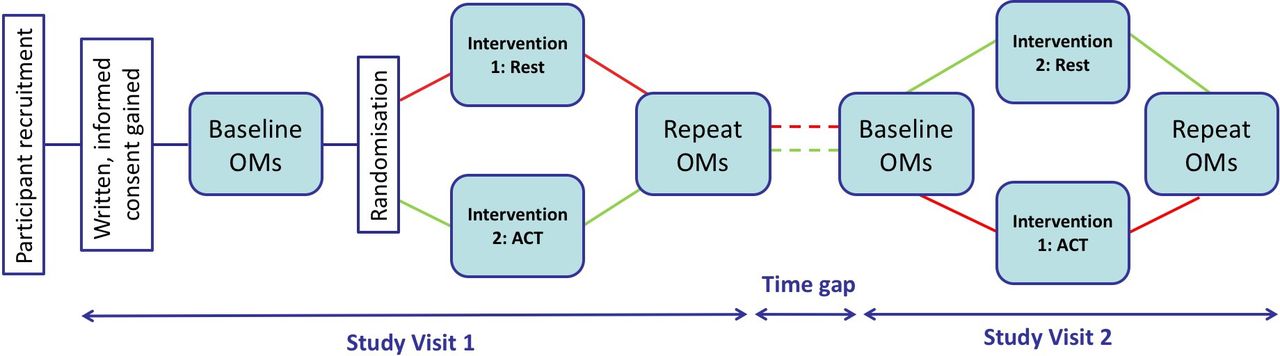
Schematic of trial design. Diagram illustrating the research journey for a trial participant, from their recruitment and giving of informed consent to completing study day one: baseline outcome measures (OMs) followed by randomisation to intervention (rest or airway clearance therapy (ACT)) and postintervention OMs. The time gap in-between visits should be no longer than 3 months. On study day 2, the participant completes the same OMs before and after the other intervention. Participation in the study is then complete.
Recruitment
Adults (16+ years) with CF registered as RBH patients are eligible for inclusion. Patients are approached during routine clinic visits for a project introduction and are given a patient information sheet (PIS) (first recruitment contact). They are given at least 48 hours to consider the PIS before a follow-up communication is completed to discuss any questions regarding study involvement and to arrange their first study visit if applicable.
Inclusion/exclusion criteria
The study’s inclusion and exclusion criteria are listed in table 1. In addition, before the start of any planned study visit (visit 1 or 2), participants are screened through discussion with the research team to see if they are unwell or if they have had any treatment changes to their CF or CF-related conditions within 4 weeks of the visit date. If treatment changes have occurred or the person is unwell, the visit is postponed.
Inclusion and exclusion criteria
Informed consent
Every potential participant is provided with a PIS, verbal study explanation and the opportunity to ask questions regarding participation at the first recruitment contact. Following this, each participant is given a full explanation of what the study entails at the start of visit 1, and has the opportunity to ask questions before the consent form is provided. Written informed consent for participation is gained at start of visit 1, and verbally confirmed at visit 2.
Study visits
Study visits are booked in advance with the participant’s agreement and consist of two separate visits to the RBH RCRF, ideally occurring within 3 months of each other. Participants complete their usual morning routines before visit attendance, which may include nebulisers and ACT, and are specifically asked to ensure they complete exactly the same routine before the second research visit.
Prior to starting study assessments, subjects are questioned to ensure they are in a stable state of their disease and continue to satisfy the inclusion and exclusion criteria. To ensure stability in disease state in between the 2 assessment days, spirometric measurements of FEV1 and FVC are required to be within 5% absolute percentage points of the recorded prevalues at visit 1 when taken at visit 2.
The format of a trial visit is shown in figure 2. Subjects perform MBW, IOS and spirometry, then a 2 min EIT recording is performed. The EIT belt stays in situ throughout the intervention period (ACT or rest) that is supervised by a specialist CF physiotherapist. Coughs, expectoration frequency and oxygen saturations are recorded during the intervention period and a 2 min EIT recording is completed. At the end of the intervention period, the participant completes a Likert scale questionnaire asking how the intervention completed has affected their breathing and sputum clearance (online supplemental appendix 1). At the end of the intervention period, a further 2 min EIT recording is completed before removing the EIT belt. MBW, IOS and spirometry are repeated immediately after the intervention period. Patient views on the OMs in terms of comfort, ease to perform and preference are gathered via a second questionnaire at the end of each study visit (online supplemental appendix 2).

Flow diagram of a study visit. CF, cystic fibrosis, EIT, electrical impedance tomography; ISO, impulse oscillometry; MBW, multiple breath washout.
Interventions
Intervention 1: airway clearance
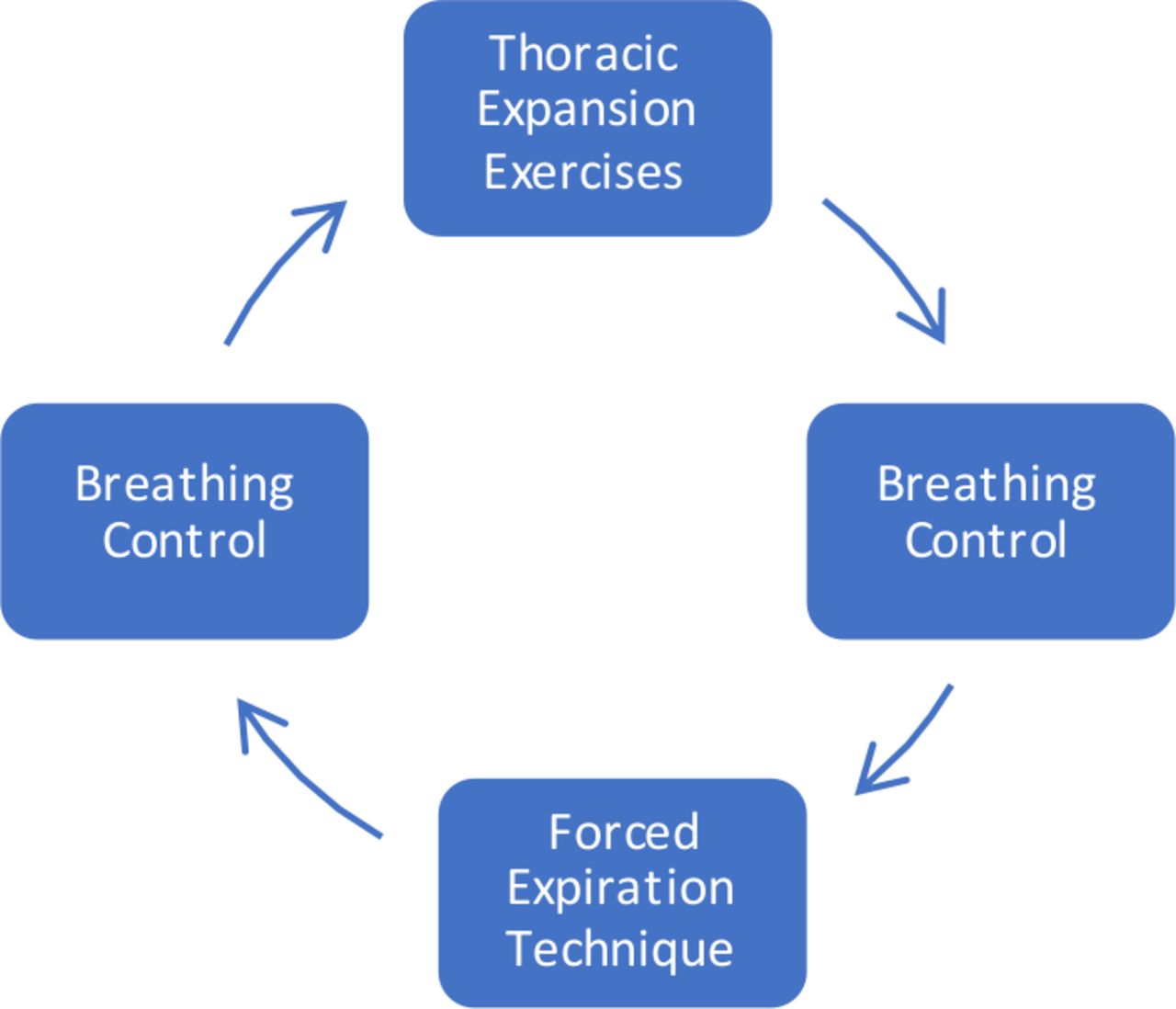
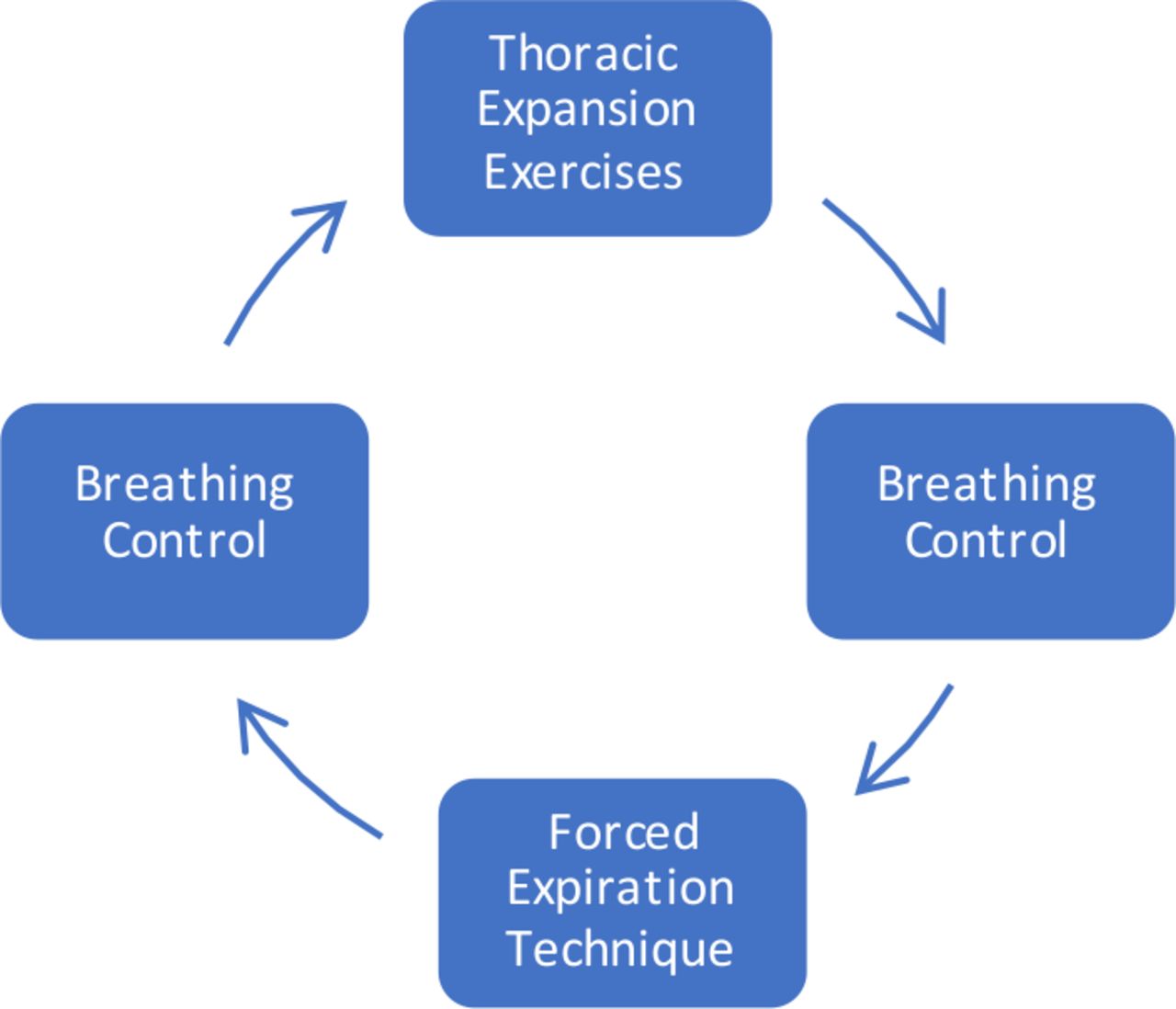
The ACBTs have been selected as the ACT within this study as it was the most common non-equipment-reliant ACT performed in the UK at the time of trial set-up,43 and is easy to learn. All participants complete ACBT as the ACT intervention to prevent complications of analysis from possible differing effects of different ACTs. ACBT includes three elements—breathing control, thoracic expansion exercises and the forced expiration technique (also known as a ‘Huff’) (figure 3).44 Teaching or revision of ACBT from a specialist CF physiotherapist is provided at the start of both trial visits to ensure correct technique. The participant completes ACBT cycles in a supported sitting position. Supervision from a specialist CF physiotherapist is provided during the intervention session to encourage good ACBT and patient motivation.
{kind=link}
{kind=link}
{kind=link}
The active cycle of breathing techniques (adapted from the International Physiotherapy Group-Cystic Fibrosis (IPG/CF) Blue Booklet, 201944).
Intervention 2: rest
In the second intervention period, the participant rests in a seated position (the same position as ACT is completed in). This intervention acts as the control arm for the study. Supervision is provided to ensure the participant does rest.
Participants are their own controls and are randomly assigned to the order that they perform the study sessions by a computerised randomisation programme. The randomisation code is held by the principle investigator who is not involved with study visits. One visit involves a period of rest for 30 min in-between assessments; the other visit involves a session of ACT using ACBT (figure 1). The length of the ACT and rest intervention periods are the same, and are predetermined prior to the first visit based on the length of the participant’s usual ACT routine. Sessions last for a minimum of 30 min, up to 60 min maximum.
To minimise bias, all OMs are completed by the same investigator as far as possible; they are blinded to the order of the study visits and are not present during the intervention period. Both the intervention periods are supervised by the specialist CF physiotherapist. The specialist CF physiotherapist and the participant are instructed to not divulge which intervention has been completed to the investigator or to any other party. The questionnaire asking how the participant feels after the intervention (online supplemental appendix 1) is given by the specialist physiotherapist and is returned to the investigator in a sealed envelope to be opened at the end of the data collection period for all subjects.
Study OMs
A summary of OM procedures is stated in table 2.
Details of outcome measures
Multiple breath washout
The MBW test is completed following the standard operating procedure for the Exhalyzer D machine (EcoMedics AG, Switzerland).45 Calibration for environmental conditions, flow and volume is completed prior to test completion. Disposable antibacterial filters and sterile mouthpieces are used. Distraction is provided during the test via a television programme. The MBW test is completed when three acceptable measurements are obtained. Once the tests are completed, the data are downloaded and analysed using Spiroware V.3.1 (EcoMedics AG, Switzerland).
Impulse oscillometry
Calibration for ambient conditions, volume and impedance reference is performed before each use. The IOS tests are completed in an upright seated position, following a standard RBH hospital protocol which was described by Paredi et al.24 Disposable antibacterial filters and sterile mouthpieces are used. The patient is asked to place their hands over their cheeks to prevent wobble and a nose clip is worn. The patient is instructed to breathe normally on the mouthpiece and at least 30 s of ‘normal breathing’ is completed before the test is commenced. Once started, the patient continues to breathe normally for a further 30 s, which is recorded. Each test is reviewed after completion and abnormal breaths (eg, coughs or swallows) excluded from the final analysis. The test is repeated when three acceptable measurements have been completed.
Spirometry
Spirometry is completed using a MicroLab spirometer (Carefusion, UK) and following the European Respiratory Society/American Thoracic Society guidelines.11 Disposable antibacterial filter mouthpieces are used for each patient. Each test is repeated three times and the best value was recorded. Percentage predicted values are obtained using the Global Lung Initiative 2012 equations.46 Calibration of the equipment is performed before each use.
Electrical impedance tomography
EIT recordings are made via electrodes connected to a thoracic belt, which is then connected via a trunk cable to the Pulmovista 500 machine (Draeger Medical UK, UK). Belt size is determined using a guide measure (Draeger Medical UK, UK). The belt is placed around the chest as per the manufacturer’s guidance (instructions for use—Pulmovista 500, Draeger, UK) at the height of the 4th–6th intercostal space with the belt moistened using water for enhanced contact. A reference electrode is attached to the seated patient using a disposable adhesive ECG electrode (Leonhard Lang GmbH, Austria). System checks and signal quality are checked prior to the first recording as per the manufacturer’s guidelines. Recordings are completed, before and after the intervention period and one in the middle of the intervention period; each recording is 2 min in duration. Recordings are uploaded to the EIT Data Analysis Tool (Draeger Medical UK, UK). Further analysis is then completed using Excel.
Oxygen saturations
Oxygen saturations are monitored continuously throughout the intervention periods using a Pulsox-300i oximeter and finger probe (Stowood Medical, UK). Recordings are analysed using Visi-Download software (Stowood Scientific Instruments, UK).
Sputum collection
Patients are given a sputum pot with lid (Unisurge, UK) to expectorate into throughout the intervention period. The pot is weighed pre and post use using portable scales (Kenex, UK).
End of trial and follow-up
There are no follow-up contacts for participants after completion of visit 2. The trial will end once 64 participants have been recruited, and have completed both study visits.
Primary and secondary outcomes
As this study assesses the five OMs with equal weight, the main measurement derived from each of the OM tests are all considered to be primary outcomes (table 3), and were all considered in the calculations to derive the optimal sample size. Other measurements that can be derived from each of the OMs will be investigated as secondary outcomes.
Primary and secondary outcome measures
Sample size calculations
In this study, we are assessing changes in OMs before and after ACT. To do this, for each individual participant we are calculating the change in the OM before and after ACT and comparing this with the change in the same OM after a period of rest. Due to the lack of agreement and definition as to what a minimal clinical important difference is for most of these OMs within the CF population,47 48 we conducted sample size calculations based on detecting minimum clinically relevant differences (MCRDs) from expert opinion and previous research findings.30 34 Sample size calculations were based on detecting MRCDs with 80% power and a significance level of 5% for all five OMs and on the premise of using a two-sample paired means test. All sample size calculations were conducted in STATA V.13.
From the calculations, FEV1 required the largest sample size of 32 participants. Initially we planned to study patients based on three severities of lung disease by FEV1 (mild (>70% FEV1 predicted), moderate (40%–70% FEV1 predicted) and severe (<40% FEV1 predicted)); therefore, we aimed to recruit 32 patients in each group. Allowing for drop outs, we aimed to recruit a total of 106 participants. However, due to difficulties recruiting participants with low lung function, the protocol was changed (with Research Ethics Committee (REC) approval in March 2020) to study two groups (FEV1 over 60% and FEV1 60% or below), requiring 70 participants.
Data analysis plan
Demographic and outcome numerical data will be summarised as mean (SD) or median (IQR) depending on the distribution of the data and categorical data will be presented with frequencies (percentage).
Between visit differences in the change in OM’s will be compared using the paired t test or the Wilcoxon sign-ranked test as appropriate. All tests will be two-sided and significance will be set at p<0.05.
The clinimetric properties of the OMs will be assessed and minimally important changes (MICs) to participants will be calculated.49 Construct validity as a measure of responsiveness will be assessed using a construct approach. Reliability will be assessed by calculating intraclass correlation coefficients and limits of agreement using Bland and Altman plots, taking measurement error between study days into account.
MICs differ from MCRDs, because MICs are calculated from patient feedback, and so represent what needs to change in an OM for a patient to feel a difference,49 as opposed to an MCRD, which is defined by clinical experts.50 MICs will be calculated using an anchor-based method49–51 derived from the results of the intervention effect questionnaire (online supplemental appendix 1).
The clinimetric properties of the secondary OMS will be explored using the same approach. Exploratory analysis of associations between the OMS and change in OMs will be completed.
To ensure blinding of investigators, no interim analysis is planned. Data analysis will be completed once full recruitment is accomplished and data gathered.
Ethics and dissemination
Ethics approval was gained for this trial from the London–Chelsea REC on 5 August 2016, with Health Research Authority (HRA) approval granted on 31 August 2016 (reference 16/LO/0995) and local research and development approval on 7 September 2016.
Safety reporting
Data on serious adverse events (SAE: as described by good clinical practice guidelines) and adverse events (AEs) will be collected during the trial visits and during time gaps in-between visits. All SAEs will be reported to the sponsor within 48 hours of notification, and will be followed until resolution. SAEs in this study include participant hospitalisation, including elective admissions, for any condition including pre-existing conditions. There are no expected AEs for this trial, except for possible fatigue from completion of interventions, and so all AEs are treated as unexpected, discussed with the trial management team, then reported to the sponsor in the annual progress report.
Changes to trial after commencement
The study details reported here are described within the trial protocol V.4.2 (date: 20 January 2020). There have been two protocol amendments, the first of which added the intervention effect questionnaire into the trial visits (online supplemental appendix 1). The first amendment was granted ethical approval by the London–Chelsea REC on 16 March 2017 and HRA approval on 27 March 2017. The second protocol amendment changed from studying three groups of participants by level of FEV1 (FEV1 mild (over 70% FEV1 predicted), moderate (40%–70% FEV1 predicted) and severe (under 40% FEV1 predicted)), to studying two groups (FEV1 over 60% and FEV1 60% or below) due to recruitment difficulties in the severe lung function group. The second amendment also included the addition of slow vital capacity into the protocol as a secondary OM. The second trial amendment was granted ethical approval by the London–Chelsea REC and the HRA in March 2020.
Publication and dissemination
All participants will receive a lay summary of results on study completion. Trial results will be presented at national and international conferences, and local scientific meetings of Imperial College, London, and RBH. A report on results will be submitted to the trial funder (National Institute for Health Research). Publication will be pursued in a relevant peer-reviewed scientific journal.
Acknowledgments
Thanks to Penny Agent; the Royal Brompton Hospital (RBH) adult cystic fibrosis physiotherapy team including Fiona Cathcart, Zelda Beverley, Tom Tobin and Paul Wilson; the Lung Clearance Index (LCI) Core Facility based at RBH especially Christopher Short and Claire Saunders; Martyn Biddiscombe; Sally Meah and the RBH research office for supporting this study.
References
Footnotes
Contributors GS, NJS, DB, OU, WB, JD and MJ were involved in the original protocol writing, review and amendments. GS and NJS wrote this protocol version for publication, with final review and comments from all authors. SC was involved in the writing and review of the statistical analysis plan.
Funding This work is funded by a Health Education England and National Institute for Health Research (NIHR) clinical doctoral research fellowship for Gemma Stanford (reference number CDRF-2014-05-055). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Competing interests JD reports other from Algipharma AS; Bayer AG; Boehringer Ingelheim Pharma GmbH & Co. KG; Galapagos NV; ImevaX GmbH; Nivalis Therapeutics; ProQR Therapeutics III B.V.; Proteostasis Therapeutics; Raptor Pharmaceuticals; Vertex Pharmaceuticals (Europe) Limited; Enterprise; Novartis; Pulmocide; Flatley; Teva and grants from CF Trust outside the submitted work. NJS reports personal fees from Vertex; Chiesi; Teva; Roche; Pulmocide and Zambon outside the submitted work.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.