Article Text

Abstract
Introduction Cigarette smoking and oxidative stress are common risk factors for the multi-morbidities associated with chronic obstructive pulmonary disease (COPD). Elevated levels of advanced glycation endproducts (AGE) increase the risk of cardiovascular disease (CVD) comorbidity and mortality. The enzyme fructosamine-3-kinase (FN3K) reduces this risk by lowering AGE levels.
Methods The distribution and expression of FN3K protein in lung tissues from stable COPD and control subjects, as well as an animal model of COPD, was assessed by immunohistochemistry. Serum FN3K protein and AGE levels were assessed by ELISA in patients with COPD exacerbations receiving metformin. Genetic variants within the FN3K and FN3K-RP genes were evaluated for associations with cardiorespiratory function in the Subpopulations and Intermediate Outcome Measures in COPD Study cohort.
Results This pilot study demonstrates that FN3K expression in the blood and human lung epithelium is distributed at either high or low levels irrespective of disease status. The percentage of lung epithelial cells expressing FN3K was higher in control smokers with normal lung function, but this induction was not observed in COPD patients nor in a smoking model of COPD. The top five nominal FN3K polymorphisms with possible association to decreased cardiorespiratory function (p<0.008–0.02), all failed to reach the threshold (p<0.0028) to be considered highly significant following multi-comparison analysis. Metformin enhanced systemic levels of FN3K in COPD subjects independent of their high-expression or low-expression status.
Discussion The data highlight that low and high FN3K expressors exist within our study cohort and metformin induces FN3K levels, highlighting a potential mechanism to reduce the risk of CVD comorbidity and mortality.
- COPD ÀÜ mechanisms
- systemic disease and lungs
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
Do lower levels of fructosamine-3-kinase (FN3K) expression confer increased risk of cardiovascular disease (CVD) in chronic obstructive pulmonary disease (COPD)?
Different levels of FN3K expression occur in subjects which is affected by smoking and disease status with potential genetic linkage to increased CVD risk.
Screening for FN3K may help to identifying those most at risk of future CVD as well as open up new therapeutic opportunities to reduce the risk of CVD and mortality in COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) extends beyond the lung and is associated with other diseases, such as cardiovascular and metabolic diseases.1 Apart from known common risk factors, such as cigarette smoking, many of the underlying molecular mechanisms driving these comorbidities remain to be investigated. Oxidative stress is a major factor in the pathogenesis of COPD2 and cigarette smoking affects both systemic inflammation and oxidative stress.3 Cigarette smoke contains damaging molecules in the form of highly reactive carbonyls commonly referred to as carbonyl stress or advanced glycation endproducts (AGEs), that have been linked to, and can accelerate, diabetic complications, such as cardiovascular disease (CVD).4
An increase in oxidative stress as a result of mitochondrial dysfunction in skeletal muscle may also increase the risk of developing type 2 diabetes mellitus through increased insulin resistance and subsequent hyperglycaemic.5–7 Moreover, mitochondrial dysfunction, which is accompanied with ROS production and an altered metabolic profile by raising ribose levels, also occurs in the lungs8 and skeletal muscle9 of COPD patients.10 Together this can lead to the development of ‘carbonyl stress’’ both in the lungs and systemically with an increase in AGEs and subsequent protein modification2 11 similar to that seen in diabetics.12
The recently identified enzyme, fructosamine-3-kinase (FN3K) and it is homologue FN3K-RP, can limit the production of AGEs from reducing sugars, such as ribose, by making the intermediate products unstable, liberating the native unmodified protein.13 Moreover, two recent publications implicate FN3K in maintaining NRF-2 function14 and in regulating histone carbonylation by ribose15 with important epigenetic consequences, both of which can impact on cell function. Both isoforms of FN3K (FN3K and FN3K-RP) are constitutive expressed throughout the human body with the heart and vasculature showing the highest levels of expression (Human Protein Atlas and GTEx Portal). Furthermore, variation within the FN3K-RP gene has been associated with CVD risk traits.16
We previously reported that high FN3K expression in diabetic patients was associated with a lower incidence of CVD risk factors suggesting that FN3K may be protective.17 Moreover, a recent study by Sartore et al18 have shown similar findings. During acute exacerbations of COPD (AECOPD), blood sugar levels and insulin resistance worsens because of the increased physiological stresses during acute illness and increased use of corticosteroid therapy.19 Such patients are treated with drugs, such as metformin, that increase insulin sensitivity and help lower blood sugar and corresponding AGE levels.20 Consequently, metformin has been shown to reduce the risk of cardiovascular disease21 as a result of lowering AGE levels.22 23
Due to the apparent role of FN3K in reducing the levels of AGEs and the potential risk of developing CVD we hypothesised that FN3K may play an important molecular link for the comorbidity associations between CVD and COPD. In order to test this hypothesis, we performed a pilot analysis of FN3K protein expression in two separate clinical studies, comprised stable COPD (study 1) and AECOPD (study 2), as well as in a chronic smoking model of COPD. In study 1 we looked at the expression of FN3K in non-smokers, smokers and stable COPD. Whereas, in study 2 we assessed the impact of metformin on FN3K expression in AECOPD, a condition where blood sugar levels and insulin resistance are known to rise. This was then complemented by a study to determine if variants in the FN3K and FN3K-RP genes are associated with a measure of cardiovascular function in COPD patients.
Materials and methods
Human study subjects
Study 1: all subjects provided informed consent and were recruited from the Section of Respiratory Medicine of the University Hospital of Ferrara, Italy undergoing lung resection for a solitary peripheral neoplasm. Thirty-six subjects in total consisted of 12 non-smokers with normal lung function, 12 smokers with normal lung function and 12 smokers with moderate (GOLD2) stable COPD. All subjects were free from preoperative chemotherapy and/or radiotherapy and had not been treated with bronchodilators, theophylline, antibiotics, antioxidants and/or glucocorticoids in the last month before surgery. The resected lung tissue samples were fixed in paraffin and then blinded in a randomised manner before examination by immunohistochemistry. This study was part of a larger project examining the molecular pathophysiological mechanisms of COPD.
Study 2: following informed consent, 23 patients with AECOPD and diabetes admitted to St George’s Hospital (London) were treated with either metformin or placebo in a randomised double-blind manner (16 treated with metformin and 7 treated with placebo). Serum samples collected on entry (baseline), at discharge (7 days after entry) and during follow-up (28 days after discharge) were then tested for FN3K protein expression by ELISA. This study formed part of a larger study investigating the impact of metformin treatment in AECOPD patients (clinical trial # NCT01247870) and full details regarding patient recruitment, exclusion criteria and dosing regimens are published elsewhere.24
All pulmonary function tests were performed as previously described.25 Both clinical studies conformed to the Declaration of Helsinki with the work being approved by the respective institutional ethics committees.
As part of the patient and public involvement policy, patients generously provided tissue samples for this study following informed consent as detailed previously. The patients and public were not involved in the study design, conduct, dissemination of results and evaluation of the studies.
Animal studies
Eight-week-old C57BL/6J wild type mice were exposed to either cigarette smoke or filtered air for 6 months as described previously.26 All exposures were approved by the local ethical committee at Rochester University, USA for work on animals and abided by the Declaration of Helsinki. Following the exposure, the animals were culled and FN3K expression was determined in peripheral lung tissue by immunohistochemistry on slices of paraffin fixed lung tissue.
Immunohistochemistry
Immunostaining of formalin fixed paraffin-embedded peripheral lung tissue was performed as previously described.27 Both human and murine FN3K were detected with a rabbit polyclonal anti-FN3K Antibody (catalogue no. AP7083a) purchased from Abgent (Cambridge, UK) at a dilution of 1.667 µg/mL and 2.5 µg/mL, respectively. Positively stained cells were counted in at least five different fields of view and expressed as a percentage of total cells for each subject or mouse.
ELISA (for FN3K and carbonyl levels)
FN3K levels in serum was assessed by ELISA using a commercial FN3K sandwich-based ELISA kit (catalogue no. CSB-EL008760HU) manufactured by Cusabio Corp and purchased from Stratech Scientific (Newmarket, UK). Serum samples were diluted 1:5 in assay diluent and processed as per manufacturer’s instructions.
Carbonyl (advanced glycated endproducts) levels were assessed in serum as per manufacturer’s instructions using the OxiSelect protein carbonyl ELISA kit (catalogue no. STA-310) purchased from Cambridge Bioscience (Cambridge, UK).
FN3K/FN3KRP genetic association study
Regression-based association testing was undertaken to identify associations of 252 FN3K and FN3KRP single nucleotide polymorphisms (SNPs) with cardiovascular and respiratory exercise performance as measured by the six-minute walk test distance (6MWD) as a percentage of predicted.28 The study population included an independent cohort of 1827 non-Hispanic Whites (NHW) and 428 African Americans (AA) defined as at-risk smokers with and without COPD from the NHLBI (National Heart,Lung and Blood Institute) Subpopulations and Intermediate Outcome Measures in COPD Study (SPIROMICS). These subjects had whole-genome genotyping data complemented by imputation resulting in the genotyping of 252 SNPs within the 34 kB genomic region encompassing FN3K and FN3K-RP genes on chromosome 17. This culminated in the representation of 18 independent, tagging SNPs in NHW and 54 SNPs in AA (r2<0.5) within FN3K and FN3KRP.
Statistical analysis on FN3K protein expression data
The statistical analysis was performed using Graph-pad Prism V.7 software. Serum FN3K expression data and carbonyl level data for AECOPD subjects treated with or without metformin were tested for normality at all data points using the Shapiro-Wilk normality test. Differences between groups (placebo vs metformin) were analysed using Tukey’s two-way analysis of variance (ANOVA) followed by unpaired t-test (Holm-Sidak method) for comparison between groups at each sampling point. To determine significant differences within each group (either placebo or metformin) with multiple comparisons between each time point, one-way ANOVA with Tukey’s post hoc analysis was undertaken. For immunohistochemical data, the Kruskal-Wallis test was applied to determine significant differences in FN3K expression between non-smokers, smokers and stable COPD. Mann-Whitney U test was applied for comparison of FN3K expression between air and cigarette smoke exposed mice. Probability values of p<0.05 were considered significant. To determine qualified comparisons on categorical data for the numbers of subjects allocated as high frequency FN3K expressers between the three groups (non-smokers, smokers and COPD) a χ2 analysis was performed.
Results
FN3K expression is reduced in COPD lung
Immunohistochemical analysis of FN3K expression in human peripheral lung tissue indicated that FN3K was expressed in airway epithelial cells (figure 1). The level of expression of FN3K across all three groups (non-smokers, smokers and stable COPD) could be separated into two distinct clusters within each group. This was determined using a mathematical model of K means cluster analysis. Across all three groups (non-smokers, smokers and COPD) the two clusters that were identified could be defined into those that had a low frequency (<15%) of FN3K positive airway epithelial cells and those that had a high frequency (≥15%) of FN3K positive airway epithelial cells (figure 1D). Further analysis revealed that the 15% cut-off was at least two SD away from the means of all samples allocated to clusters with a low frequency of FN3K positive cells within each group. In keeping with this definition, in healthy non-smokers, 83% (10 of 12) of subjects expressed very little FN3K with only 2.2%±1.4% of airway epithelial cells expressing FN3K. Whereas, 17% (2 of 12) of non-smokers expressed FN3K with 66%±7% of epithelial cells staining positive for FN3K. In contrast, 75% (9 of 12) of smokers were positive for FN3K expression exhibiting a significant (p<0.05) increase in FN3K expression with 31.3%±6.3% of epithelial staining positive for FN3K compared with 2.0%±1% of epithelial cells expressing FN3K in the 25% of smokers defined as low FN3K expressers. In the stable COPD group, the number of subjects still exhibiting widespread expression of FN3K fell to 33% (4 of 12) with 50%±11% of epithelial cells expressing FN3K. Consequently, 67% (8 of 12) of the COPD subjects had significantly lower (p<0.05) levels of FN3K expression with only 4.1%±2% of epithelial cells expressing FN3K.
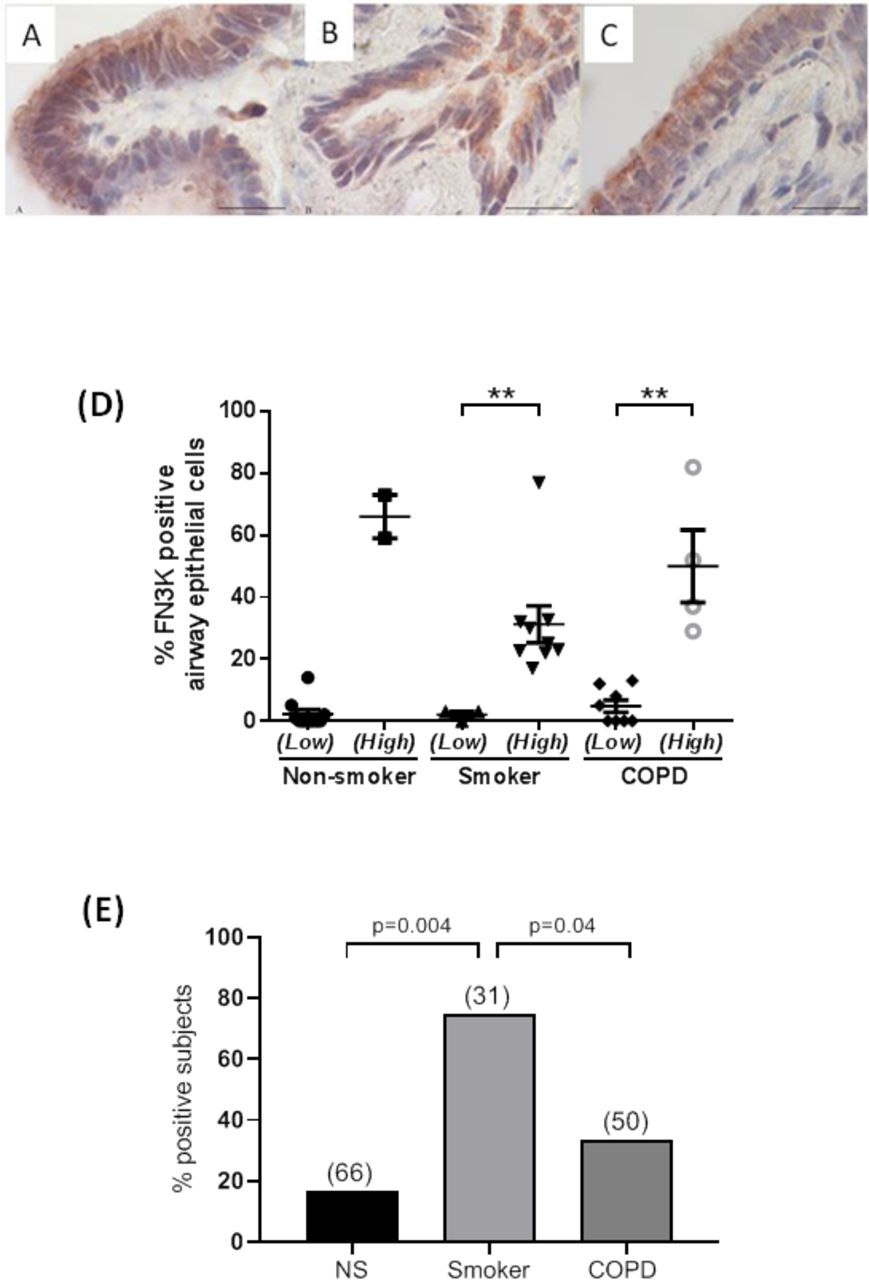
Representative immunohistochemical staining for fructosamine-3-kinase (FN3K) in bronchiolar epithelial cells of stable chronic obstructive pulmonary disease (COPD) patients and control subjects (A: non-smoking control; B: control smoker with normal lung function; C: patients with stable COPD; 100× magnification; bar: 20 µm). (D) The percentage of FN3K immunopositive epithelial cells from peripheral lung sections following immunohistochemical staining. Each data point is the average score of immunopositive epithelial cells counted over several random fields of view for each subject. The mean±SEM is overlaid on top of the scatter plot for each cohort sub grouping. Each cohort (non-smoker, smoker with normal lung function (smoker) and COPD) is split into two subgroups, those exhibiting a low frequency of FN3K expression (low) and those exhibiting a high frequency of FN3K expression (high). Non-parametric Kruskal-Wallis test was employed, where applicable, to determine significant differences within each cohort subgrouping (**p<0.01). (E) The percentage of subjects exhibiting immunopositive FN3K expression in >15% of lung epithelial cells, as illustrated in(D), between non-smokers (NS), smokers with normal lung function (smokers) and stable COPD patients (COPD). The numbers in brackets represent the mean percentage of FN3K immunopositive epithelial cells in each cohort exhibiting high FN3K expression. A χ2 test was used to check for independence. The probability of independence (p) between the three groups is illustrated.
Figure 1E illustrates the impact of these results by expressing the percentage of subjects in each group expressing high levels of epithelial FN3K expression. A χ2 analysis was performed to examine the relationship in the number of subjects identified as high frequency FN3K expressers between all three groups (non-smokers, smokers and COPD). This demonstrated that that there was a significant association between smoking and high levels of FN3K expression when compared with non-smokers (χ2 (1, N=24)=8.22; p=0.004) and COPD (χ2 (1, N=24)=4.20; p=0.04). These results would imply that smoking induces expression of FN3K in airway epithelial cells, presumable as a protective mechanism in order to combat the increased levels of carbonyl stress/AGEs that are present and formed by cigarette smoke.4 However, the ability to maintain FN3K expression and therefore a protective mechanism to combat the increased levels of carbonyl stress or AGEs in lung epithelial cells appears to have been lost in a number of subjects with stable COPD. Interestingly, in those subjects with stable COPD that still maintained an elevated level of FN3K expression, there was a more widespread expression of FN3K with more epithelial cells (50%) staining positive for FN3K compared with smokers with 33% of lung epithelial cells expressing FN3K.
Metformin induces increased expression of FN3K in AECOPD
In study 2 involving 23 subjects with AECOPD and diabetets, metformin significantly (p<0.0001) enhanced systemic FN3K expression over a 35-day period from initial trial entry to follow-up following discharge (figure 2A). In contrast, those subjects given placebo (figure 2B) showed no significant change in systemic FN3K expression over the same time period. Furthermore, this increase in FN3K expression in the metformin treated group was significantly (p<0.05) higher than the levels observed in the placebo group at both the discharge and follow-up sampling points. We have previously shown that fructosamine levels were significantly reduced in the metformin treated group from trial entry to follow-up, unlike the placebo group which showed no significant reduction.24 Further analysis here revealed that systemic levels of carbonyl were also significantly (p<0.001) reduced by 61% at follow-up compared with trial entry in the metformin treated subjects (figure 3). Carbonyl levels also significantly (p<0.01) fell over the same time period in the placebo group, but only by 50%. The 11% greater fall in carbonyl levels in the metformin group compared with the placebo group at the follow-up time point just failed to reach significance (p=0.08).
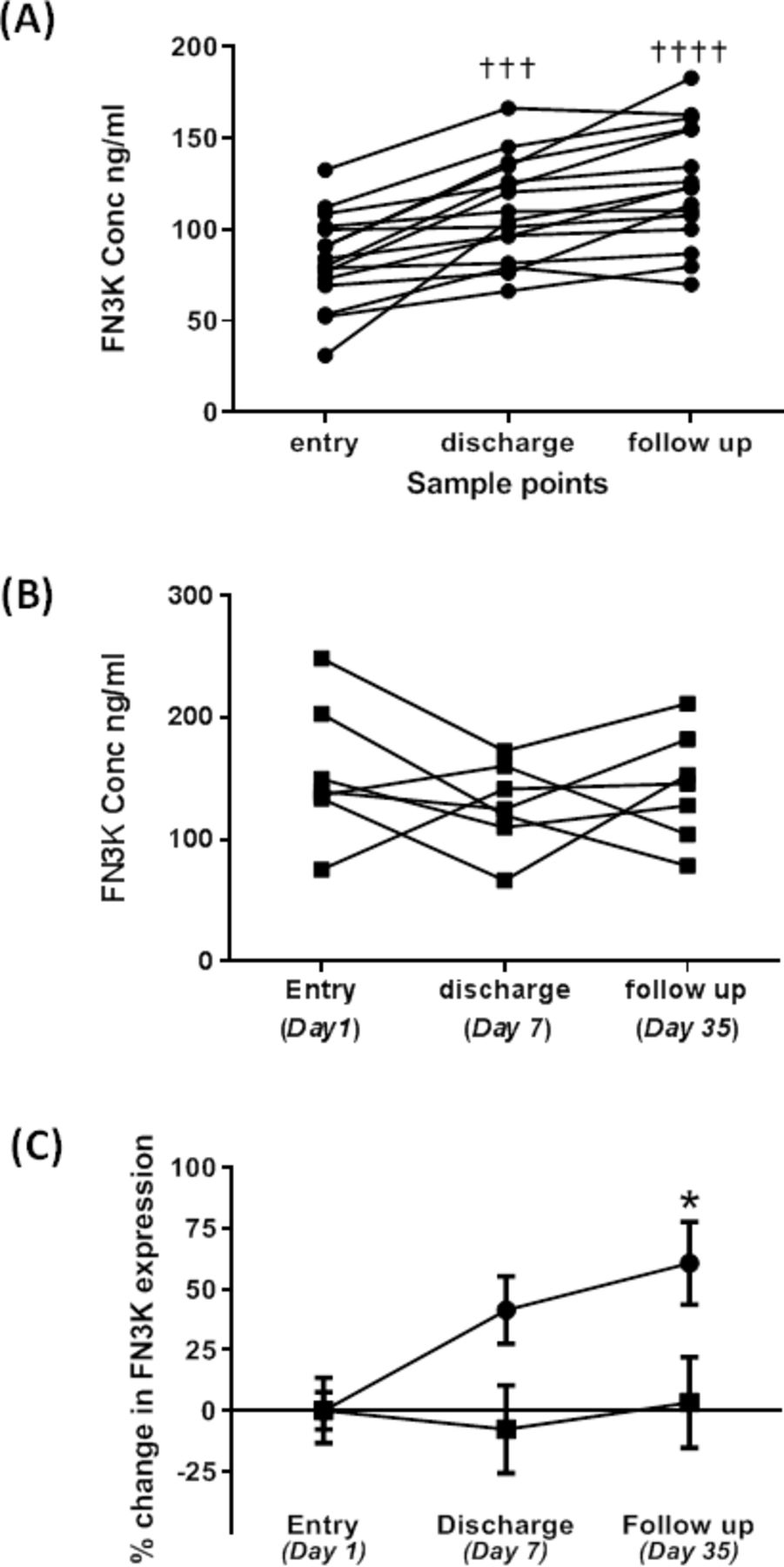
Impact of metformin (A) versus placebo (B) on serum fructosamine-3-kinase (FN3K) levels in chronic obstructive pulmonary disease (COPD) patients with an acute exacerbation. Data are plotted as the mean±SEM for FN3K protein expression as assessed by ELISA at each of the sampling points. Entry was at day 1, with a median discharge at day 7 (days 4–10) and follow-up at day 35. Data are expressed as ng/mL FN3K (A and B) and per cent change from baseline entry (C). In (A) and (B) one way analysis of variance (ANOVA) and Tukey’s multiple comparison test was used to determine significant changes relative to entry within each group (either placebo or metformin. A p<0.05 was considered significant (†††p<0.001, ††††p<0.0001). (C) Statistical analysis was performed using two way ANOVA and Sidak’s multiple comparison test across both placebo and metformin groups to test for significant differences between placebo (∎) and metformin (●) at each sampling point. A p<0.05 was considered significant (*p<0.05).
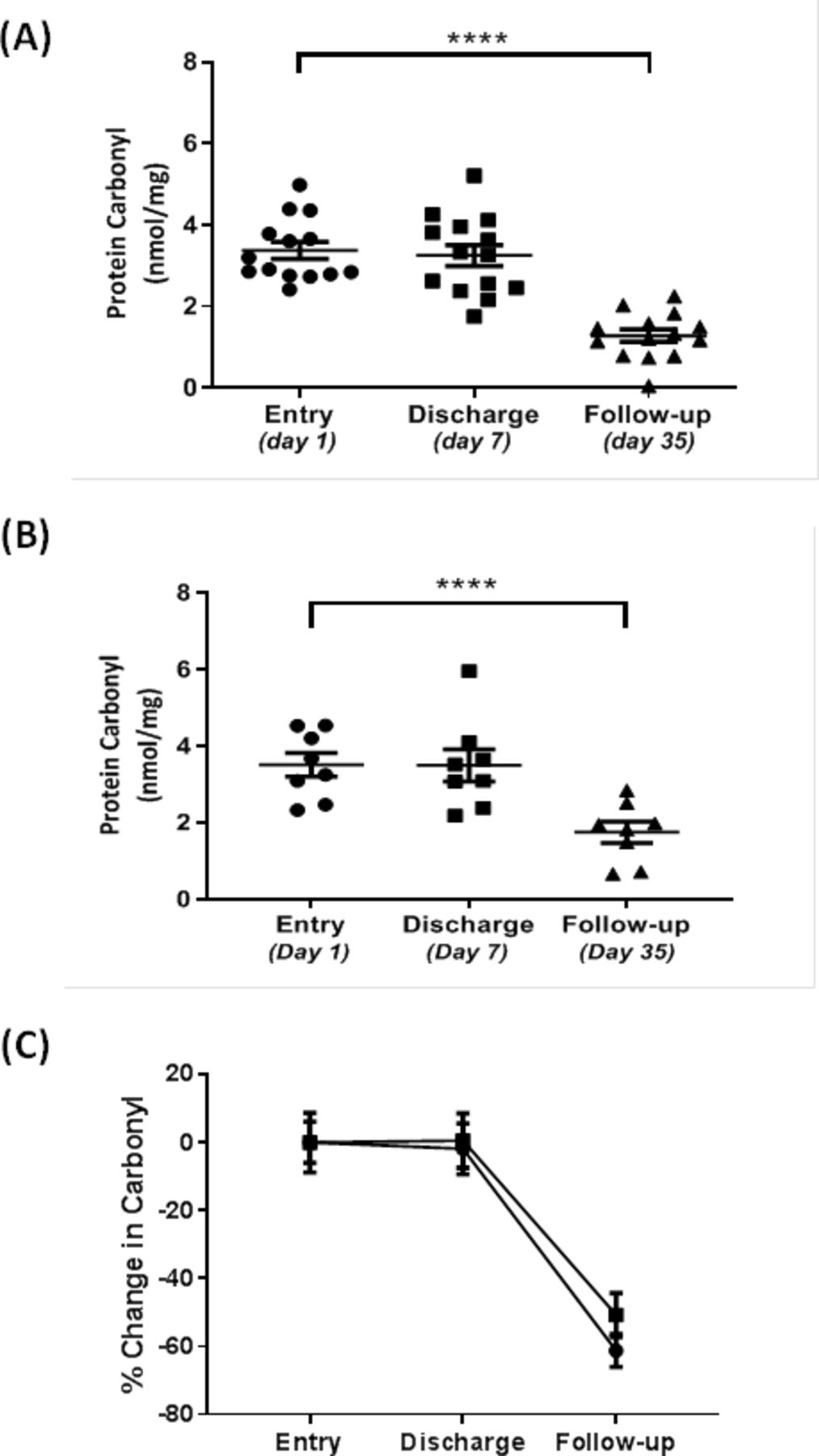
Impact of metformin (A) versus placebo (B) on serum protein carbonyl levels in chronic obstructive pulmonary disease (COPD) patients with an acute exacerbation as measured by ELISA. Data are plotted as a scatter plot with mean±SEM (A and B) and per cent change from baseline entry (C). Placebo is represented by solid squares and metformin treatment by solid circles in (C). Entry was at day 1, with a median discharge at day 7 (days 4–10) and follow-up at day 35. To test for significant difference, one way repeated measures analysis of variance was performed with Tukey’s post hoc analysis. A p<0.05 was considered significant (****p<0.0001).
FN3K expression is reduced in the lungs of cigarette smoke exposed mice
In order to ascertain whether the reduction in FN3K expression in COPD lung was a disease related event, we looked to see if it could be replicated in an animal model of COPD. Using mice that were chronically exposed to cigarette smoke for 6 months and exhibiting many of the pathological features of COPD as previously reported,26 we looked at expression of FN3K in peripheral lung tissue by immunohistochemistry. Figure 4 shows that in mice, following a 6-month exposure to cigarette smoke there was a significant reduction (p<0.05) in FN3K expression in airway epithelial cells compared with mice exposed to filtered air over the same time period. This suggests that over a long period of time, cigarette smoke may be causing a reduction in FN3K protein expression, either through increased proteolysis or through a negative impact on gene expression at the epigenetic or transcriptional levels.
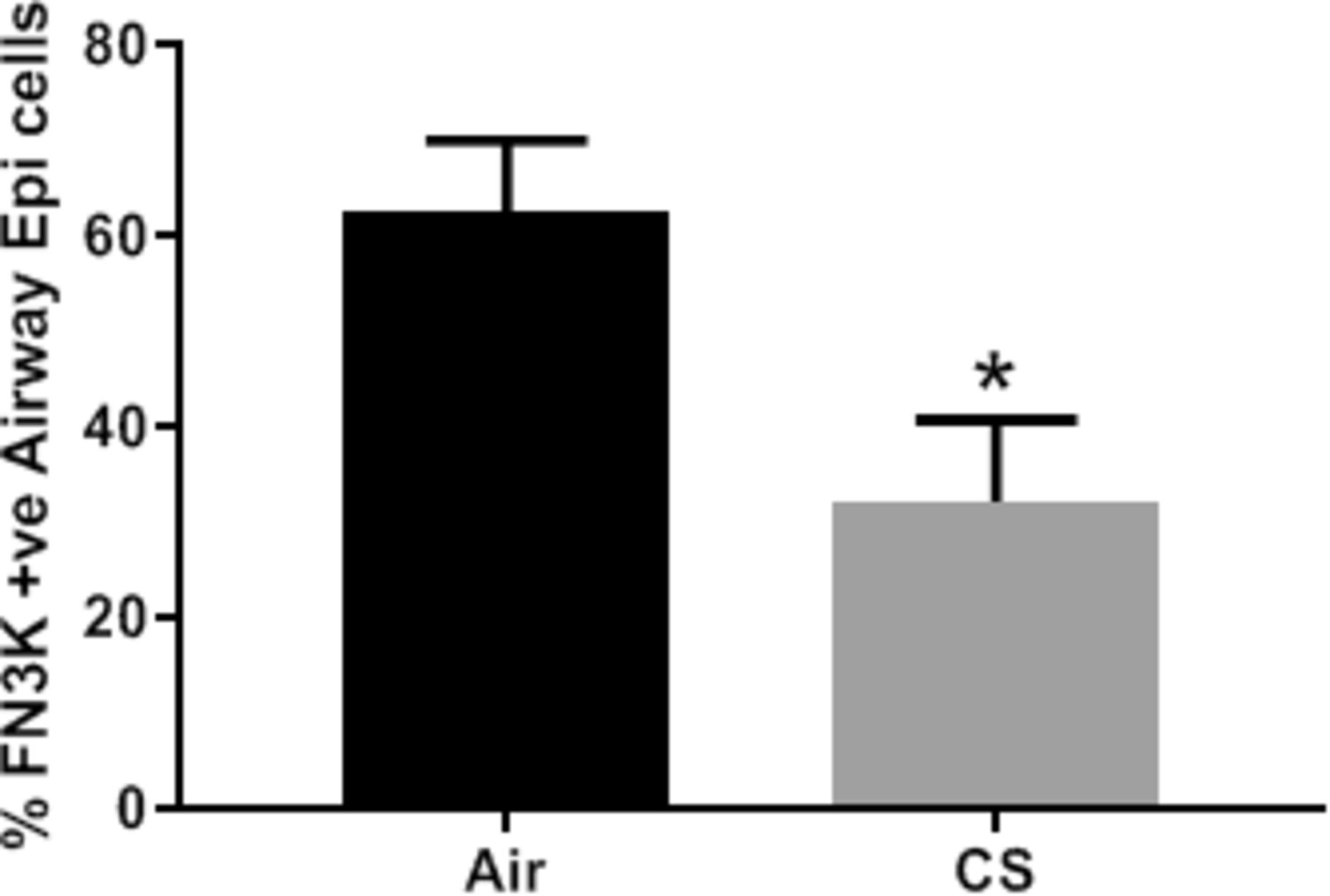
Fructosamine-3-kinase (FN3K) expression is reduced in murine airway epithelial cells exposed to cigarette smoke for 6 months. C57BL/6J wild type mice were exposed to cigarette smoke (CS) or ambient air (Air) for 6 months the percentage of airway epithelial cells staining positive for FN3K expression in peripheral lung sections following immunohistochemical staining was recorded over several random fields of view for each mouse and the data expressed as the mean±SEM (N=8 for each group). Further details are in the Materials and methods section. Statistical significance was determined using Mann-Whitney U test (*p<0.05).
Association of FN3K SNPs in COPD with cardiovascular function
Table 1 illustrates the top five independent nominal SNP associations with per cent predicted 6MWD (p=0.008 to 0.02) in the SPIROMICS data set. However, these associations did not meet the significance threshold for multiple comparisons (p=0.0028 in Whites, p=9.3×10−4 in AA). Moreover, the top five SNPs were not replicated in independent cohorts from COPDGene, GENKOLS, ECLIPSE or NETT individually or with meta-analysis (p>0.05). Nevertheless, one SNP on FN3K-RP (rs1046875) tagged for two additional SNPs (rs1046896 and rs1046917) that were all previously associated with increased HbA1c and a corresponding increased risk of coronary artery disease.29 30
Top genetic associations of FN3K and FN3KRP tagging single nucleotide polymorphisms with six-minute walk distance (6MWD) in non-Hispanic Whites from Subpopulations and Intermediate Outcome Measures in COPD Study (MAF = Mean Allele Frequency)
Discussion
In this pilot study, FN3K protein was detected both in the lungs and systemically in COPD. The levels of FN3K positive (FN3K+) epithelial cells in non-smoker, smoker and COPD airways separated into two distinct phenotypes: very low or high numbers of FN3K+ positive cells. However, a similar separation was not seen in the smaller number of COPD subjects in the metformin study. This separation of FN3K+ subjects into two distinct groups is similar to that reported in diabetic subjects.17 Moreover, these phenotypes correlated with cardiovascular risk, whereby low FN3K expression was associated with a larger G-gap and greater cardiovascular risk and vice versa.17
The accumulation and increased presence of AGEs or carbonyl stress along with the corresponding pathophysiological mechanisms are well documented31 and a known risk factor for developing CVD.23 In diabetes the formation of AGEs is linked to high glucose levels and the presence of oxidative stress through a process known as glycation. However, in COPD high levels of carbonyl stress are also present as a result of chronic inflammation, oxidative stress2 and elevated ribose levels resulting from metabolic reprogramming.10 Ribose is 20 times more reactive than glucose in glycating proteins and forming AGE/carbonyl adducts.32 Consequently, both COPD and diabetes are influenced by inflammation and oxidative stress resulting in increased carbonyl stress and AGEs although they are driven through different aetiological stimuli (figure 5). Increased carbonyl stress and AGEs have been linked to CVD23 as well as many other comorbidities associated with COPD.33 As FN3K is widely expressed throughout the body, it is plausible that FN3K may be functioning as a protective clearance mechanism to remove the damaging precursors to AGE formation and that low levels of FN3K expression will lead to increased CVD risk (figure 5). In support of our hypothesis is the observation that in control smokers with normal lung function, we observed an increase in the numbers of cells expressing FN3K protein within the lower airways, presumably to protect from the increase in carbonyl stress and AGE levels. However, this protective mechanism would appear to be impaired in COPD as the numbers of FN3K+ cells in the lung were reduced. Furthermore, this downregulation in the numbers of FN3K+ cells was modelled in a chronic smoking model of COPD. This might suggest that pathophysiological mechanisms prevalent in COPD to trigger downregulation in FN3K expression are being replicated in the chronic cigarette smoke exposure animal model used here. We have previously shown that both oxidative stress and carbonyl levels are elevated in the chronic smoke exposure model.34 Clearly further investigation is warranted into how chronic exposure to oxidative stress/cigarette smoke can lead to down regulation in FN3K expression. Consequently, as a result of our findings reported here, the chronic smoking model may provide a suitable platform for further investigations. Indeed, little is known about the mechanisms controlling FN3K expression but a complex interaction between SNPs within the FN3K locus, inducible protective mechanisms upregulating FN3K expression and activity in response to cigarette smoke and epigenetic changes may exist.35 It is also likely that oxidative stress may have a direct impact on FN3K activity. However, a limitation in the findings reported here was that there was insufficient sample to measure FN3K enzymic activity. To try and address a possible linkage between FN3K SNPs and CVD risk in COPD, we analysed the genotyping data from the SPIROMICS cohort consisting of at-risk smokers and COPD subjects. 6MWD is a measure of cardiorespiratory function determined by multiple factors that when evaluated in the context of the CVD risk factors determined by FN3K /FN3K-RP (body mass index, haemoglobin A1C, systolic blood pressure) it precludes these loci as genetic determinants for 6MWD.16 While 6MWD was the only data available to us at the time within this study cohort, a more reliable marker of CVD risk could be evaluated using more detailed phenotypes of CVD risk factors in COPD cohorts, such as MRI-calcium scores, lipid profiles, haemoglobin A1C or CT scan-based measures of adiposity in conjunction with multi-omic (transcriptomic/epigenomic) approaches. Consequently, our negative findings for FN3K and FN3K-RP variation in relation to 6MWD does not exclude an indirect contribution leading to increased CVD risk and comorbidity. Indeed, one of the SNPs identified in our study (rs1046875) is linked to variation associated with increased CVD risk traits.29 30

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fructosamine-3-kinase (FN3K) as a potential gatekeeper to prevent the accumulation of damaging AGEs that may predispose subjects to increased risk of cardiovascular disease. Accumulation of AGEs and carbonyl stress may come directly through raised uncontrolled blood glucose levels in diabetes or indirectly through chronic oxidative stress from cigarette smoking in COPD resulting in mitochondrial dysfunction, an altered metabolic profile and elevated cellular ribose levels and increased carbonyl stress. AGE, advanced glycation endproduct; COPD, chronic obstructive pulmonary disease; CVD, cardiovasculardisease.
Given that increased ribose levels can accrue in COPD subjects as a result of an increase in the pentose phosphate pathway10 and poorly controlled diabetics also exhibit increased ribose levels, both of which can lead to increased AGE and corresponding increased CVD risk.6 It is plausible that FN3K-RP genetic variation results in alterations in ribose derived AGE products subsequently linked to CVD risk. While the case is not fully proven here in COPD subjects, it does offer the possibility that there may still be a linkage, although indirectly, between FN3K polymorphisms and CVD risk in COPD subjects. Indeed, SNPs in FN3K have been strongly associated with changes in sRAGE,36 which in turn has been linked to CVD risk.37 However, a recent study by Sartore et al18 has reported that a cluster of FN3K SNPs when expressed together were associated with increased risk of microvascular and macrovascular complications. It is possible therefore that no single direct linkage between FN3K variation and CVD may exist but instead requires interactions between different FN3K polymorphisms, and possibly RAGE expression as well, to confer increased risk to CVD within COPD subjects.
Metformin administration to COPD patients with AECOPD significantly increased serum FN3K levels over the course of an exacerbation following admission into hospital irrespective of the basal level. In contrast, serum FN3K levels did not change with placebo. This increase was mirrored by a corresponding fall in carbonyl (AGE) levels from entry into the study to follow-up in both the placebo and metformin groups. Although there was a 10% difference in carbonyl levels between the two groups, this just failed to reach significance (p=0.08). This was in contrast to that seen for fructosamine levels in the same study where metformin, but not placebo, caused a significant fall in serum fructosamine levels.24 It is possible that metformin could not have a significant effect on carbonyl levels in our study, as the exacerbation and subsequent production of carbonyls had already begun before metformin was administered. Indeed, in a study by Dallak et al38 when metformin was administered prior to the induction of a diabetic state subsequent increases in carbonyl (AGE) levels were prevented. This failure to reach significance may reflect the importance of other unknown mechanisms that aid the resolution of the exacerbation episode or that this study was not sufficiently powered to detect these changes. Indeed, a potential major limitation for the two pilot studies involving stable COPD and AECOPD reported here were the low number of subjects involved. However, post-hoc analysis of statistical power revealed that both studies achieved 100% power to discriminate significant differences (p<0.001) in FN3K expression between the different cohorts under investigation. Metformin has been shown to decrease CVD risk and resultant mortality in both diabetics and non-diabetics.14 39 40 It is unclear how metformin’s impact on FN3K expression may affect pathophysiological mechanisms and subsequent comorbidity risk of CVD in COPD patients, although Dallak et al have shown that metformin can reduce levels of AGEs.38 Several pathophysiological mechanisms such as endothelial dysfunction and increased coagulopathy within the vasculature are associated with increased risk of CVD in COPD.41 Moreover, AGEs have been implicated in vascular stiffness, atherosclerotic plaque formation, thrombogenesis, increased coagulopathy, vascular calcification and endothelial dysfunction with consequential impacts on the risk of mortality.42 Therefore, the ability of FN3K to remove fructosamine and ribosamine residues on modified proteins and thereby reduce the carbonyl load and AGE formation may have a clinical impact on these pathologies and survivability. Metformin can also directly lower AGE levels by reacting with AGE adducts containing dicarbonyls,43 in particular methyl glyoxal,44 a highly reactive dicarbonyl and a by-product of increased glycolysis,45 a pathway that has been shown to be elevated in COPD.10 Given that the manipulation of upstream metabolic events involving fructosamine will only impact on subsequent AGE formation, and the removal of existing ‘long-lived’ AGE/ carbonyls may take considerably more time, a longer time course may therefore be needed to observe whether changes in FN3K expression would result in notable changes in basal AGE/carbonyl levels and in turn clinical outcome. The limitations of the present assay in measuring all carbonyls may also be inappropriate, and more specific assays detecting unique AGE/carbonyl adducts known to be associated with increased CVD risk may be more appropriate. However, a recent study by Ho et al46 indicated that metformin had a beneficial impact on survivability in COPD patients with diabetes which may be attributed to the metformin-induced increase in FN3K expression reported here, thereby reducing AGEs and subsequent mortality.
In conclusion, this pilot study shows that the distribution and expression of FN3K protein in COPD and age-matched control subjects (both non-smokers and smokers with normal lung function) appears to fall into one of two groups, those with low levels of FN3K protein or those with high levels of FN3K protein expression. Moreover, smoking appears to trigger a protective mechanism whereby FN3K protein levels are increased, but this is apparently lost in most but not all COPD subjects. Furthermore, metformin elevates FN3K expression which can reduce carbonyl stress (AGEs) thereby potentially reducing the risk of developing CVD. Clearly further work needs to be done to demonstrate that a clear mechanistic link is present between carbonyl stress, FN3K expression and CVD risk in COPD, with or without diabetes, and potentially in ageing where systemic AGE levels are also known to rise. Finally, this study highlights a potential mechanistic explanation for the benefits of providing metformin to lower CVD and other disease risks associated with increasing AGE levels in COPD.
Acknowledgments
The authors would like to thank the SPIROMICS participants and participating physicians, investigators and staff for making this research possible. More information about the study and how to access SPIROMICS data are at www.spiromics.org. We would like to acknowledge the following current and former investigators of the SPIROMICS sites and reading centres: Neil E Alexis, MD; Wayne H Anderson, PhD; Mehrdad Arjomandi, MD; Igor Barjaktarevic, MD, PhD; R Graham Barr, MD, DrPH; Lori A Bateman, MSc; Surya P Bhatt, MD; Eugene R Bleecker, MD; Richard C Boucher, MD; Russell P Bowler, MD, PhD; Stephanie A Christenson, MD; Alejandro P Comellas, MD; Christopher B Cooper, MD, PhD; David J Couper, PhD; Gerard J Criner, MD; Ronald G Crystal, MD; Claire M Doerschuk, MD; Mark T Dransfield, MD; Brad Drummond, MD; Christine M Freeman, PhD; Craig Galban, PhD; MeiLan K Han, MD, MS; Nadia N Hansel, MD, MPH; Annette T Hastie, PhD; Eric A Hoffman, PhD; Yvonne Huang, MD; Robert J Kaner, MD; Richard E Kanner, MD; Eric C Kleerup, MD; Jerry A Krishnan, MD, PhD; Lisa M LaVange, PhD; Stephen C Lazarus, MD; Fernando J Martinez, MD, MS; Deborah A Meyers, PhD; Wendy C Moore, MD; John D Newell Jr., MD; Robert Paine, III, MD; Laura Paulin, MD, MHS; Stephen P Peters, MD, PhD; Cheryl Pirozzi, MD; Nirupama Putcha, MD, MHS; Elizabeth C Oelsner, MD, MPH; Wanda K O’Neal, PhD; Sanjeev Raman, MBBS, MD; Stephen I Rennard, MD; Donald P Tashkin, MD; J Michael Wells, MD; Robert A Wise, MD; and Prescott G Woodruff, MD, MPH. The project officers from the Lung Division of the National Heart, Lung, and Blood Institute were Lisa Postow, PhD, and Lisa Viviano, BSN.
References
Footnotes
Contributors AA, CR and PC undertook the experimental work. VEO and JLC provided the genetic analysis of the SPIROMICS cohort. GC, EHB, AWH and AP provided the clinical samples necessary for the studies. IR developed and provided the tissue samples from the animal model studies. GC, IA, JLC, SD and PK designed the study. All the authors help contribute to the data analysis, interpretation and preparation of the manuscript. PK was the lead investigator for this work.
Funding Genetic analysis was supported by grants from the NIH NHLBI K08 HL118128 and R01 HL142992. SPIROMICS was supported by contracts from the NIH/NHLBI (HHSN268200900013C, HHSN268200900014C, HHSN268200900015C, HHSN268200900016C, HHSN268200900017C, HHSN268200900018C, HHSN268200900019C, HHSN268200900020C), and a grant from the NIH/NHLBI (U01 HL137880, and supplemented by contributions made through the Foundation for the NIH and the COPD Foundation from AstraZeneca/MedImmune; Bayer; Bellerophon Therapeutics; Boehringer-Ingelheim Pharmaceuticals; Chiesi Farmaceutici S.p.A.; Forest Research Institute; GlaxoSmithKline; Grifols Therapeutics; Ikaria; Novartis Pharmaceuticals Corporation; Nycomed GmbH; ProterixBio; Regeneron Pharmaceuticals; Sanofi; Sunovion; Takeda Pharmaceutical Company; and Theravance Biopharma and Mylan. IA was supported by a Wellcome Trust grant# 093080/Z/10/Z, EPSRC (EP/T003189/1) and by the UK MRC (MR/T010371/1 and G1001367/1). PK was supported by a COPDMAP grant from the UK MRC (G1001367/1). EB and AH were supported by a grant from the British Lung Foundation (COPD10/7).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval Local ethics committee of the University Hospital of Ferrara, Italy. South East Research Ethics Committee (reference 10/H1102/62).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. All data relevent to the study are deidentified participant data and are included in the article. Further details are available from the corresponding author.