Article Text

Abstract
Background Nasal potential difference (NPD) and intestinal current measurements (ICM) are cystic fibrosis transmembrane conductance regulator (CFTR) biomarkers recommended to make a diagnosis in individuals with inconclusive sweat test and CFTR genetics and a clinical suspicion for cystic fibrosis (CF) or CFTR-related disorder (CFTR-RD).
Methods NPD and ICM were measured according to standard operating procedures of the European Cystic Fibrosis Society Diagnostic Network Working Group.
Results We assessed 219 individuals by NPD or ICM who had been referred to our laboratory due to clinical symptoms suggestive of CF, but inconclusive sweat test and CFTR genetics (median age: 16.3 years, range 0.4 to 76 years). CF or CFTR-related disorder was diagnosed in 22 of 29 patients (76%) with a CFTR genotype of unknown or variable clinical significance and in 51 of 190 carriers (27%) of one (35/42) or no (16/148) identified CFTR mutation. If two CFTR sequence variants had been identified, the outcome of NPD and ICM was consistent with the classification of the CFTR2 database. Moreover, a suspected false-positive diagnosis of CF was confirmed in seven and withdrawn in eight patients. Of 26 individuals assessed by both NPD and ICM, eleven individuals exhibited discordant tracings of ICM and NPD, with one measurement being in the CF range and the other in the normal range.
Conclusion The majority of patients whom we diagnosed with CF or CFTR-RD by extended electrophysiology are carriers of the wild-type CFTR coding sequence on at least one of their CF alleles. The disease-causing genetic lesions should reside in the non-coding region of CFTR or elsewhere in the genome, affecting the regulation of CFTR expression in a tissue-depending fashion which may explain the large within-group variability of CFTR activity in the respiratory and intestinal epithelium seen in this group.
- cystic fibrosis
- bronchiectasis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the impact of the improved protocols of nasal potential difference (NPD) or intestinal current measurements (ICM) to make a diagnosis in individuals with cystic fibrosis (CF)-suggestive symptoms but inconclusive sweat test and CFTR genotype?
Cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction was demonstrated in more than one-quarter of referred individuals who carry the wild-type CFTR coding sequence on at least one of their CF alleles. Individual signatures of NPD and ICM profiles associated with variable organ manifestation were characteristic for these patients with residual, but still insufficient CFTR activity.
The outcome of the CFTR biomarker study on individuals with inconclusive sweat test and CFTR genetics suggests that we can globally expect a large yet undiagnosed cohort of thousands of individuals with CFTR dysfunction who lack state-of-the-art CF care.
Introduction
The diagnosis of cystic fibrosis (CF) or cystic fibrosis transmembrane conductance regulator-related disorder (CFTR-RD) is based on the clinical interpretation of the phenotypic features and confirmed by sweat test and CFTR mutational analysis.1 2 However, a subgroup of patients with CF-suggesting illness presents with inconclusive sweat test and CFTR genetics. In this case, guidelines recommend assessment of CFTR function by alternate methods such as nasal potential difference (NPD)3 4 and intestinal current measurement (ICM).5 NPD measures epithelial sodium channel (ENaC)-mediated sodium and CFTR-mediated chloride ion conductance of nasal respiratory epithelium.6 7 ICM is an ex vivo method in which a freshly excised rectal biopsy is tested in an Ussing chamber for its CFTR-mediated chloride secretory responses to agonists that increase intracellular cyclic adenosine monophosphate (cAMP).5 8 9
NPD and ICM reliably discern the responses of subjects with classic CF and non-CF controls. Yet, historically, there has been substantial variation of reference values between laboratories. This situation has been remedied by the Cystic Fibrosis Foundation Therapeutics, Inc, Therapeutics Development Network (CFFT TDN) coordinating centre and the European Cystic Fibrosis Society Diagnostic Network Working Group (ECFS DNWG) by the establishment of standard operating procedures (SOPs).
At our clinic, NPD6 7 and ICM protocols8 9 were installed in 1998. By 2011 the in-house protocols were replaced by electronic data capture and the SOPs of the diagnostic networks. Since then, we have performed NPD and ICM in the context of ENaC and CF modifier studies, characterisation of CFTR genotypes, post-approval studies on CFTR modulators and CF diagnostics.
NPD and ICM are quantitative biomarkers of CFTR activity in the respiratory and intestinal epithelium that can be used to define a threshold between ‘CFTR-normal activity’ and ‘CFTR dysfunction’. NPD and ICM measurements in cohorts of healthy non-CF controls, patients with CF and individuals with borderline sweat test led to the definition of empirical thresholds that differentiate between CF and non-CF groups. The discriminative parameters, that is, the cumulative chloride secretory response5 for ICM and the so-called ‘Wilschanski’10 and ‘Sermet scores’11 for NPD recordings were executed according to in-house protocols.
Here we report on our experience with the SOPs released by the diagnostic networks. The SOPs were applied to the cases in CF diagnostics that are most challenging to differentiate between CF, CFTR-RD and non-CF, that is, individuals with suspected false-positive diagnosis of CF, individuals with inconclusive sweat test and a CFTR mutation genotype of unknown or variable clinical significance12 and—most frequently—individuals with CF-suggestive symptoms, but normal or intermediary sweat test values and no or only one identified disease-causing CFTR mutation.
Methods
ICM and NPD protocols
NPD measurements were initially carried out according to the SOPs 528.00 and 529.00 issued by the CFFT TDN in February 2009, and since August 2013, according to SOP NPD_EU001 of the ECFS DNWG. The European SOP uses shorter lines, does not use warmed solutions, abstains from the exploration of the potentials within the nostril in successive steps and uses a Marquat catheter which is more comfortable for the investigated subject. The SOP for NPD was slightly modified in-house. In brief, the procedure to prepare bubble-free agar bridges was optimised because bubbles in agar bridges typically drive high transition potential and impede any reliable NPD measurements. Moreover, we modified the preparation of the subcutaneous reference electrode to ensure a stable reference potential. Finally, some imperfections in the setup of stock solutions were eliminated. First, ATP stock solution was stored at −80°C to avoid depurination which occurs at relevant rates in ice clathrates at the recommended storage temperature of −20°C.13 Second, the make-up of phosphate buffers was modified so that magnesium phosphate does not precipitate, as this affects pH and the reproducibility of NPD measurements.
ICM was measured according to the SOP of the ECFS DNWG, V.2.7, which was implemented at our CF Electrophysiology Laboratory in December 2011. The SOP of the ECFS DNWG introduced some changes from the initial Rotterdam protocol developed by Hugo de Jonge and Henk Veeze.8 9 For activation of CFTR, the brominated cAMP derivative 8-Br-cAMP was substituted by the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX) and the CFTR potentiator genistein14 with the concentration of all other secretagogues being maintained. More importantly, the sequence of the addition of secretagogues was changed so that the responses to carbachol and histamine are measured in the presence of the CFTR activators forskolin/IBMX. Reference values of ICM diagnostic parameters have only been published for the Rotterdam protocol,5 but not for the ECFS DNWG SOP so far. We therefore chose to develop in-house reference values for non-CF subjects. Table 1 shows the outcome of ICM in 68 non-CF healthy controls. The cut-off value between patients with exocrine pancreatic sufficiency (PS) CF and controls was shifted from 34 µA/cm2 (original Rotterdam protocol) to 39 µA/cm2 (SOP) for the cumulative chloride secretory response ΔIsc, Forsk/IBMX, ΔIsc, carbachol and ΔIsc, histamine which has been shown to represent the most conclusive diagnostic ICM parameter to differentiate patients with questionable CF into PS CF and ‘CF unlikely’ groups.5
ICM according to SOP ICM_EU001, V.2.7 in 68 healthy non-CF control subjects
Workflow to make a diagnosis by NPD and/or ICM
Indications to perform diagnostic testing by NPD and/or ICM
The CF electrophysiological laboratory Hannover accepted requests in-house from other departments and externally from certified CF centres or from pneumologists or paediatricians in private practice who regularly take care of patients with CF. Indications to perform ICM and/or NPD were:
clinical symptoms suggestive for CF or CFTR-RD but an inconclusive sweat test in the normal or intermediary range and no or only one identified CFTR mutation (group 1),
an inconclusive sweat test and a CFTR genotype of unknown or variable clinical significance (group 2),12
a pathological sweat test but no overt clinical symptoms of CF disease (group 3),
and the re-evaluation of an existing, but questioned diagnosis of CF (group 4).
Although the congenital bilateral absence of the vas deferens (CBAVD) is the most common CFTR-RD,2 the aetiology of male infertility has never been an indication for CFTR functional analysis in the reporting period.
Referral criteria
After the first contact had been established in person or by phone or email, the referring physician was asked to substantiate his suspected diagnosis of CFTR dysfunction by providing case history and medical findings for review and to transmit the results of Gibson-Cooke pilocarpine iontophoresis sweat tests and the report of CFTR mutation analysis of a certified human genetic laboratory15 by Fax or postal service. In case of group 1, CFTR mutation analysis had to comprise the sequencing of all exons and flanking intron regions of the CFTR gene. In case of groups 3 and 4, the referring physician was asked to provide clinical and laboratory evidence of why CF was unlikely or had been falsely diagnosed.
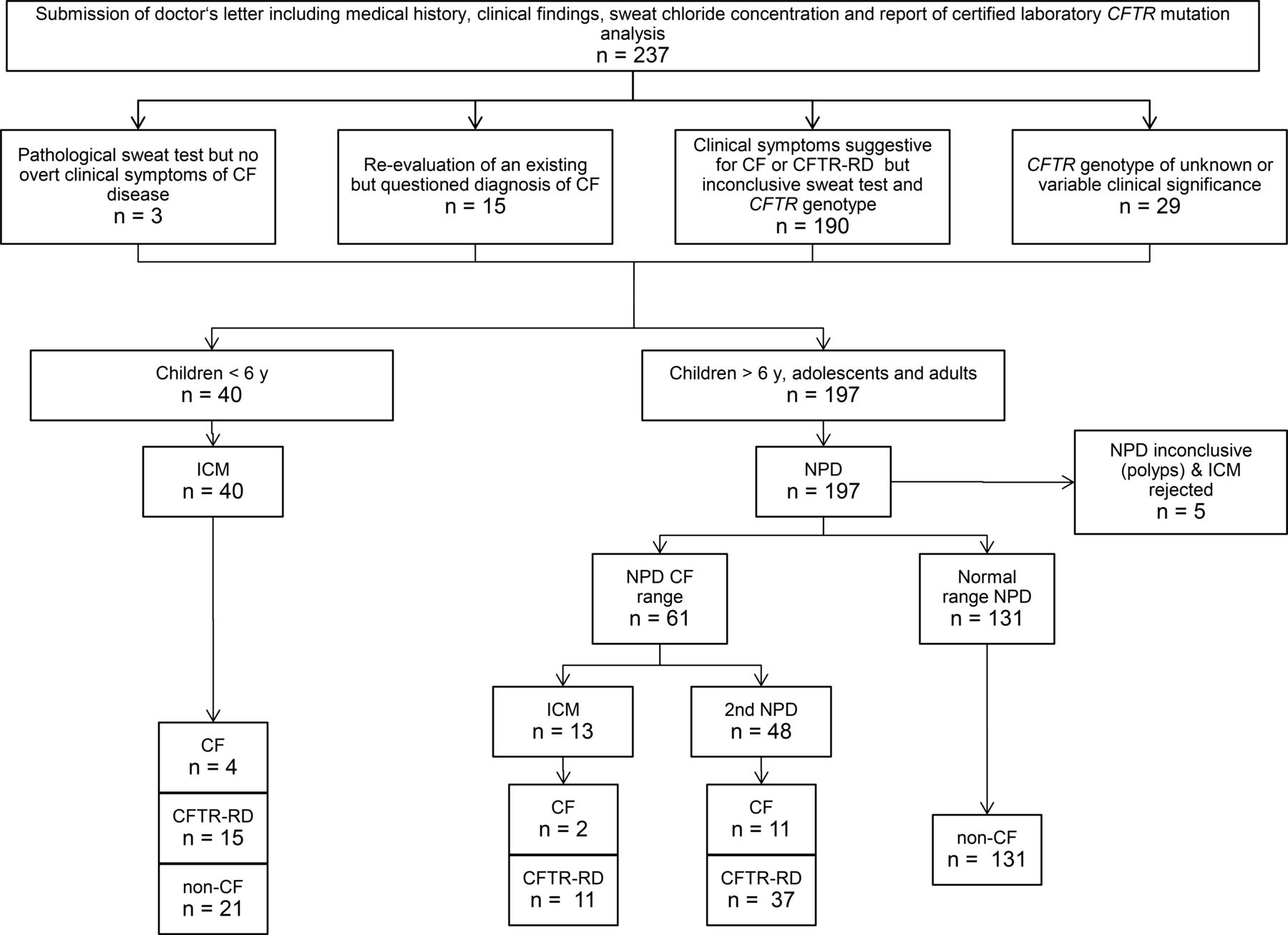
Data about sweat chloride concentration, CFTR genotype, anthropometry, exocrine pancreatic status and features of respiratory, gastrointestinal and hepatobiliary disease were then reviewed by a CF physician of the local CF team (online supplemental table S1). If the CF physician endorsed that CFTR biomarker analysis was indicated, an appointment at the CF electrophysiology laboratory was made. Small children were typically invited to undergo ICM whereas children from 6 years upward, adolescents and adults were first invited to a NPD measurement (figure 1).
Supplemental material

Flow diagram of diagnostic process. CF, cystic fibrosis; CFTR-RD, cystic fibrosis transmembrane conductance regulator-related disorder; ICM, intestinal current measurement; NPD, nasal potential difference.
Patients who systemically administered beta blockers or topically applied rhinologicals such as steroids, xylometazoline or oxymetazoline were requested to stop the medication for the 24 hours prior to the NPD assessment. If the antihypertensive therapy was considered to be indispensable, numerous appointments were scheduled in case of a pathological outcome of the NPD. Since cigarette smoke induces CFTR dysfunction,16 17 smokers were requested to refrain from smoking for at least the 2 weeks prior to the day of NPD measurement.
On-site procedures on the day of assessment
Prior to the NPD measurement, the nares were examined with a rhinoscope for mucosa disruption, oedema, erythema, polyp and secretion. NPD measurements were carried out according to SOP NPD_EU001 of the ECFS DNWG.
Prior to ICM, the bleeding time was determined. Rectal tissue was obtained with a suction biopsy device. ICM of the rectal biopsies was performed according to the SOP ICM_EU001 of the ECFS DNWG, V.2.7. The response of a biopsy to the secretagogues was only evaluated if the biopsy was viable, not leaky and not folded.
Evaluation of NPD/ICM and diagnostic decision
Members of groups 1 and 2 were suspected to be clinically affected by CFTR dysfunction according to the CF experts’ opinion, but both chloride concentration in sweat test and CFTR mutation analysis were inconclusive. Alternatively, the previous diagnosis of CF was questioned by CF experts for individuals of groups 3 and 4. These two scenarios called for a stringent procedure prior to making any decision whether or not the propositus was affected by CFTR dysfunction. Hence the NPD and ICM tracings were evaluated in a stepwise approach by first identifying the unequivocal cases with normal CFTR activity followed by the in-depth appraisal of both electrophysiological and clinical features of the remaining subgroup to make a diagnosis.
First step: If the basic potential in the NPD was less negative than −30 mV in both nares and if in at least one nare the depolarisation potential on exposure to chloride-free solution and to isoproterenol exceeded the basic potential and/or if the Sermet score11
S = −0.11 Δ (LowCl-Iso) −0.05 Δ amiloride (CF: S<0.27; non-CF: S>0.27)
was larger than 0.7, CF and CFTR-RD were excluded and the NPD was measured only once. If only subtle depolarisation potentials with chloride-free solution and isoproterenol were recorded with a Sermet score of less than 0.7 in both nostrils, an ICM was performed or the NPD was repeated on a separate day.
CF and CFTR-RD were excluded by inspection of the ICM tracings if the cumulative secretory response of at least one of the four biopsies exceeded the lower quartile of non-CF individuals of 100 µA/cm2 or if each of the four biopsies exceeded the threshold of CF/non-CF of 39 µA/cm2.
If CF and CFTR-RD had not been excluded by the above-mentioned criteria, CF, CFTR-RD and non-CF were discriminated along the 2011 guideline of the global diagnostic algorithm for CF and CFTR-RD2 wherever applicable and modified by our experience gained during the last 10 years. The evaluation differed between groups 1 and 2 and groups 3 and 4.
Patients of groups 1 and 2
A patient was classified ‘non-CF’ if the NPD of both nostrils and/or the ICM tracings of all four biopsies were consistently in the normal non-CF range.
A patient was diagnosed to be affected by CF if the Sermet score of both nostrils and/or the ICM tracings of all four biopsies were consistently in the abnormal CF range and if patient history and/or current laboratory and clinical findings revealed CF typical features in airways and gastrointestinal tract.
CFTR-RD is defined as a ‘clinical entity associated with CFTR dysfunction that does not fulfil the diagnostic criteria for CF’.2 The patient was diagnosed to be affected by CFTR-RD:
if the majority of examined biopsies in the ICM showed tracings in the abnormal CF range and the mean cumulative chloride secretory response of the biopsies was in the CF range,
and/or if the Sermet score of both nostrils was consistently in the abnormal CF range (S<0.27),
and if clinical symptoms compatible with CFTR dysfunction were present in at least one organ system, that is, hepatobiliary or gastrointestinal tract, or upper or lower airways.
Please note that according to our experience, all examined biopsies exhibited consistent and uniform ICM tracings in the non-CF range in people with non-CF and in the CF range in people with CF. Conversely, biopsies from individuals with CFTR-RD typically showed a patient-characteristic temporal profile of the chloride secretory responses to forskolin/IBMX, carbachol and histamine. The amplitude of the current response often varied between the biopsies, indicating spatial fluctuations of CFTR-mediated chloride transport in the donor tissue consistent with residual, but subnormal CFTR activity.
Patients of group 3 and 4: The diagnosis of CF was confirmed if the NPD of both nostrils and/or the ICM tracings of all four biopsies were consistently in the abnormal CF range. Alternatively, the previously made diagnosis of CF was withdrawn if the NPD of both nostrils and/or the ICM tracings of all four biopsies were consistently in the normal non-CF range.
Patient and public involvement
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
Results
Study population
From August 2009 to May 2020, 237 individuals were examined to make a diagnosis by NPD and/or ICM with electronic data capture at our CF electrophysiology laboratory (online supplemental table S1). Patients’ median age on the day of assessment was 16.3 years (inner quartiles: 9 to 32 years; range 0.4 to 76 years). ICM was mainly performed in preschool children and NPD was preferred for the assessment of children from 6 years upward, adolescents and adults (figure 1). Patients were either referred in-house or externally from other CF centres or physicians in private practice with expertise in CF care. With the exception of two ear, nose and throat specialists, all external requests were from paediatricians or adult respiratory physicians. The majority of patients showed clinical features of disease in airways and /or intestine compatible with CFTR dysfunction but both sweat test and the sequencing of all exons and flanking intron sequences of the CFTR gene had turned out to be inconclusive (n=190). No requests were obtained to resolve the aetiology of male infertility. Twenty-nine probands were carrying a CFTR genotype of unknown or variable clinical significance12 requiring further analysis by CFTR bioassay. Minor indications were the examination of children with a pathological sweat test, but no CF-typical clinical symptoms (n=3) and the re-evaluation of an existing CF diagnosis (n=15). Online supplemental table S1 displays for each individual the age at the day of assessment, clinical features and the primary data of sweat test, NPD and ICM.
Outcome of assessments
One-third of the study population (81 of 237 people) was diagnosed CF or CFTR-RD (table 2). NPD or ICM tracings in the normal range were recorded for 152 persons. No reliable ICM and NPD measurements were feasible in five subjects because of contraindications (prolonged bleeding time and haemorrhoids) or no detectable respiratory epithelium in both nostrils.
Evaluation of subjects with suspected CFTR dysfunction by ICM and NPD
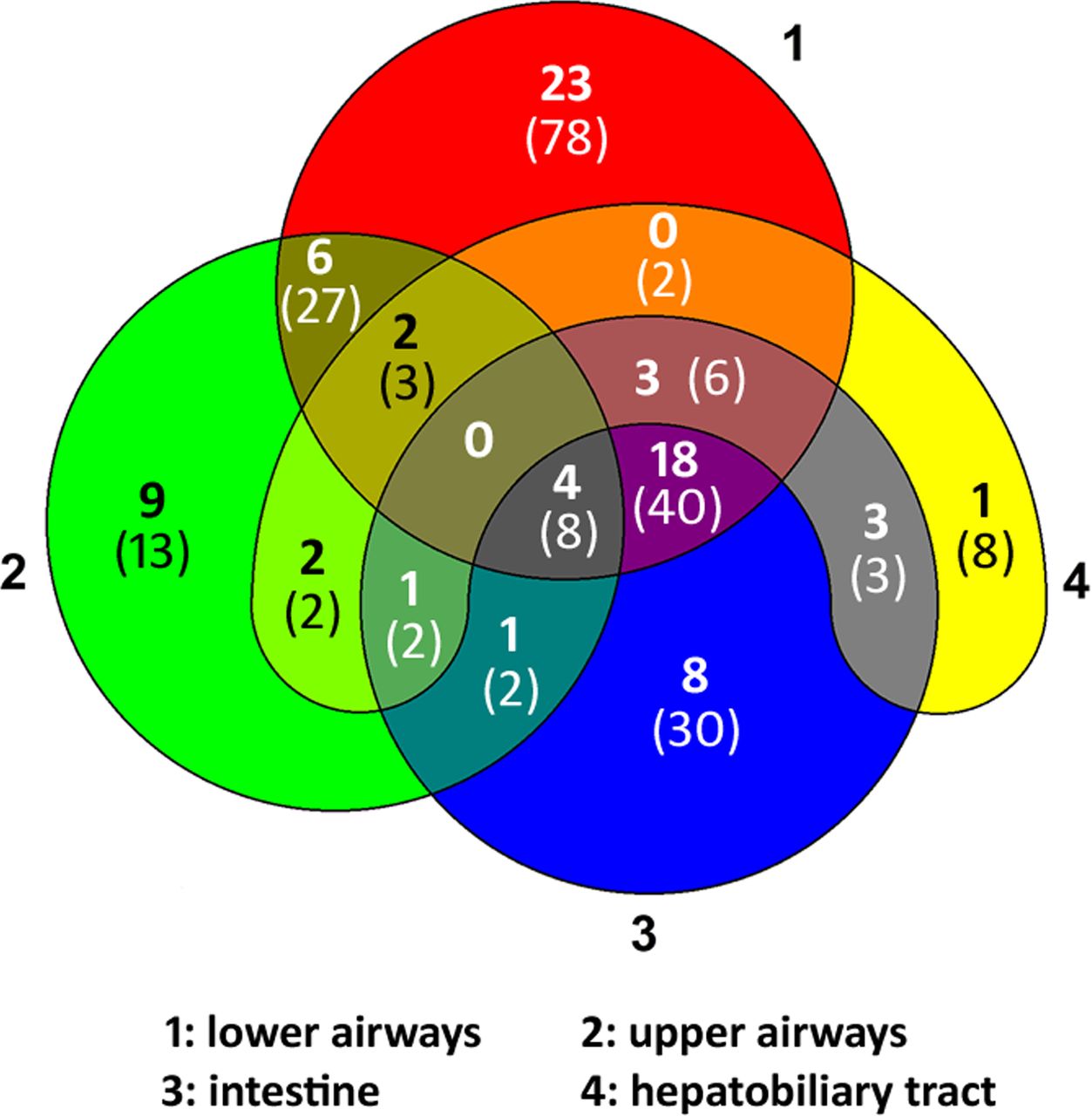
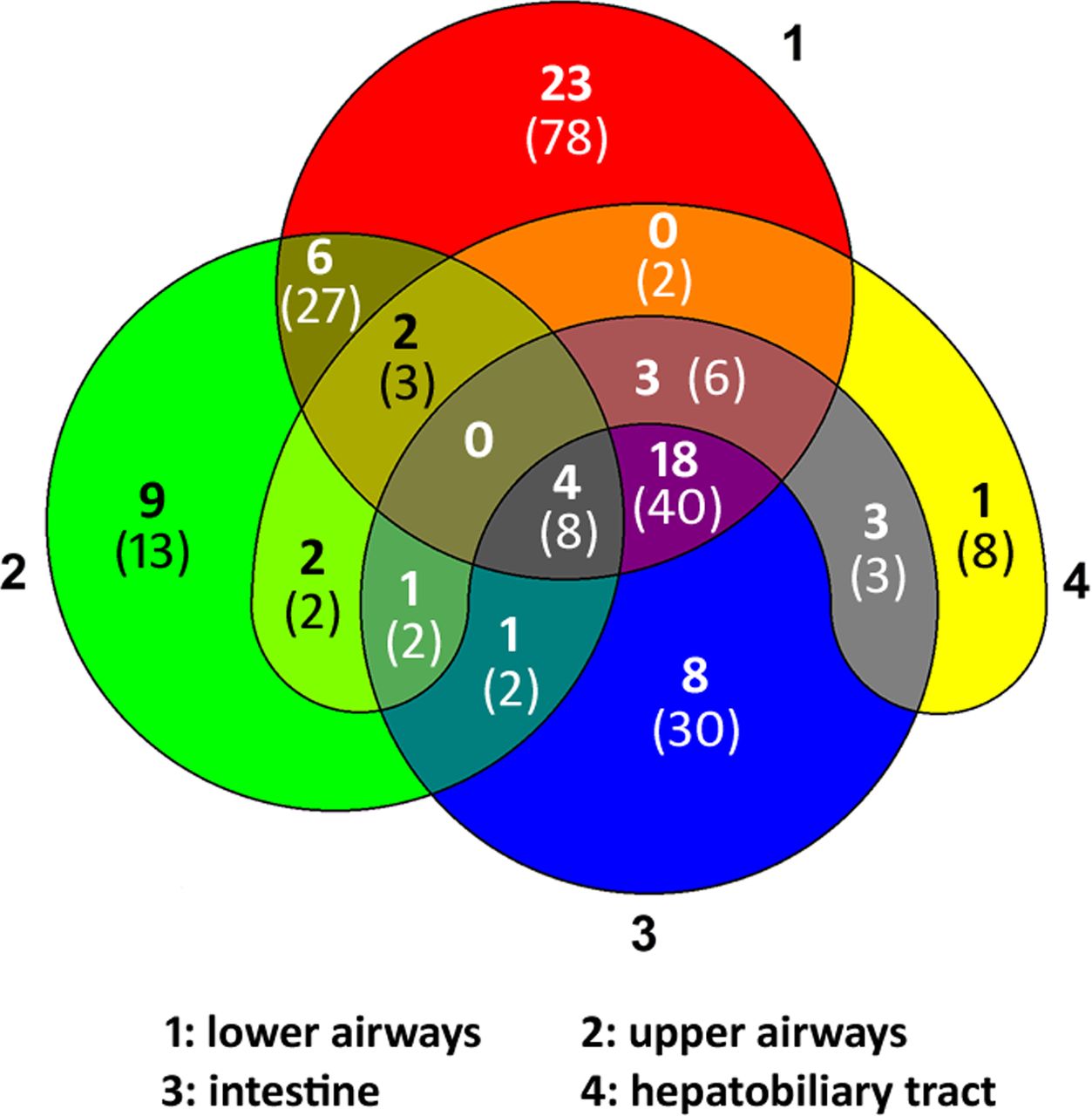
Features compatible with the disease manifestation of CFTR dysfunction could be documented for 224 of the 237 persons (online supplemental table S1). Clinical documentation was not sufficiently informative for the classification of the other 13 persons. The Venn diagram (figure 2) displays the distribution of CF-suggestive findings in upper and lower airways, intestine and hepatobiliary tract compiled from the patient’s record, the doctor’s letter and the medical history and clinical investigation performed at the day of assessment. Nobody showed the typical constellation of CF pulmonary, intestinal and hepatobiliary disease. The CF-typical combination of pulmonary disease with abdominal pain, underweight or pancreatic disease was reported by just one-sixth of the patient cohort. Conversely, more than half of the study population showed CF-compatible features in only one organ system. The highest rate of newly diagnosed patients with CF or CFTR-RD occurred among the group which solely presented upper airway symptoms such as chronic rhinosinusitis18 or nasal polyps followed by the group with CF-compatible findings in two organ systems (figure 2).
{kind=link}
{kind=link}
Distribution of clinical findings suggestive for CFTR dysfunction in 224 patients seen at the CF electrophysiology laboratory to make a diagnosis by NPD and/or ICM. The Venn diagram depicts the involvement of lower (1) and upper (2) airways, intestine (3) and/or hepatobiliary tract (4) compiled from the patient’s record, the doctor’s letter and the on-site anamnesis and clinical investigation performed on the day of assessment. The number in bold font indicates the number of individuals diagnosed by CFTR biomarker to be affected by CF or CFTR-RD; the number in brackets in normal font gives the total number of individuals in this group. CF, cystic fibrosis; CFTR-RD, cystic fibrosis transmembrane conductance regulator-related disorder; ICM, intestinal current measurement; NPD, nasal potential difference.
Both NPD and ICM were performed in 26 subjects of group 1 with clinical symptoms suggestive for CF but inconclusive sweat test and CFTR genotype (table 3). The outcome of the two CFTR bioassays was consistent for 15 persons, but inconsistent for two subgroups of seven and four persons. The first group was affected by severe pulmonary disease, but only mild intermittent symptoms of gastrointestinal disease. They produced NPD tracings in the CF range, but their ICM revealed chloride secretory responses of the intestinal epithelium in the lower normal range. The other four persons had experienced repetitive episodes of pancreatitis and abdominal pain and had been occasionally affected by upper or lower airway infections. Their ICM showed low CFTR activity in the CF range, whereas the chloride conductance of their respiratory epithelium was in the lower normal range.
Clinical features and CFTR biomarker analysis of subjects assessed by both NPD and ICM
Of the 81 newly diagnosed patients with CF or CFTR-RD, 66 patients were of German and four patients of Slavic descent. The 11 non-Caucasians had Turkish (n=6), Arabian (n=4) or African parents (n=1).
CFTR activity of individuals with CFTR genotypes of unknown or variable clinical significance
Online supplemental table S2 lists the 29 CFTR genotypes of unknown or variable clinical significance in the cohort. Twenty-one of the 29 investigated patients showed abnormal NPD and ICM tracings consistent with CF or CFTR-RD (online supplemental table S1). The recordings of the other eight subjects were in the normal range. Eleven of the 21 sequence variants are listed in the CFTR2 database.12 Compound heterozygosity for p.Leu997Phe was associated with normal ICM and NPD consistent with the classification of CFTR2 that this missense variant is non-CF causing. The missense variants p.Arg74Trp and p.Asp1270Asn were part of a complex triple allele with p.Val201Met (online supplemental table S2). The two index cases were affected by mild CF.19 Discordant CFTR biomarker phenotypes were noted for carriers of the p.Arg117His mutant. The three p.Phe508del /p.Arg117His compound heterozygotes in our cohort showed CFTR dysfunction in the CF range, whereas the p.Arg117His-7T homozygote exhibited normal CFTR activity consistent with a previous report of normal clinical features and normal CFTR activity in two homozygous p.Arg117His-7T adults.20 Ten sequence variants of our group are rare and have yet not been characterised in their CFTR activity. Six of the 10 variants were classified by the CFTR biomarkers as disease-causing in the examined individuals (online supplemental table S2).
Supplemental material
Discussion
This report presents the yet largest cohort of individuals with inconclusive sweat test and CFTR genetics who were examined by NPD and ICM to make a diagnosis. CF or CFTR-RD was diagnosed in one-third of examined subjects. This high detection rate probably reflects the fact that virtually all cases were referred to us by physicians with in-depth knowledge of CF. However, even though our relatively high detection rates may be partly attributed to the expertise of the referrers, the rate of 51 newly diagnosed CF patients with no or one mutation in the coding sequence was unexpectedly high. Prior to the setup of the reference laboratory, the German CF registry recorded just 100 newly diagnosed cases within a 10-year period who were 18 years or older at the time of diagnosis. French colleagues recently reported that the CFTR genotype remains incomplete in 1% of CF cases.21 In contrast, by sequencing all exons and flanking intronic sequences, we identified two CF-causing CFTR mutations in 100% of exocrine pancreatic insufficiency (PI) CF patients of German descent, but only in 40% to 50% of PS CF patients.22 Whole-gene sequencing of CFTR recently uncovered a high prevalence of several disease-causing deep intronic variants such as c.3874-4522A>G21 23–25 that are not amenable by the mode of CFTR testing applied to our cohort. Even if these newly identified intronic mutations were included into CFTR mutation analysis, the data of our domestic CFTR biomarker study suggest that we can globally expect a large yet undiagnosed cohort of thousands of individuals with CFTR dysfunction and non-informative CFTR genetics who lack state-of-the-art CF care. Most subjects will not suffer from typical CF, but from CFTR-RD. The disease-causing sequence variations do not necessarily reside in the CFTR gene but could alternatively be located in regulatory genetic elements of CFTR expression encoded elsewhere in the genome. The genetically unresolved patients with inconsistent outcome of NPD and ICM, or an index case with temporally fluctuating chloride concentrations in sweat test of 40, 23, 91, 22 and 78 mmol/L on five separate occasions (text in supplement) would be strong candidates of being a carrier of a genetic modifier of CFTR expression in trans.
On the other hand, if subjects were carrying two CFTR sequence variants in trans, the outcome of NPD and ICM was consistent with the classification of the CFTR2 database.12 Carriers of variants classified by CFTR2 as non-CF causing presented with NPD and ICM tracings in the normal range, whereas variants of variable clinical significance also showed variable CFTR activity in the respiratory and intestinal epithelium.
ICM and NPD are in place at our site since more than 20 years. The technical quality of the tracings has always been sufficient for the differentiation between CF PI patients and non-CF healthy controls. However, the joint efforts of the scientific community to optimise and standardise protocols and equipment, particularly the introduction of internal controls, troubleshooting and electronic data capture,3–5 11 26–30 have been instrumental for us to perform reproducible measurements with low drift and high signal-to-noise ratios during the last 12 years . The whole range of CFTR activity, particularly the broad spectrum of CFTR dysfunction in individuals with monosymptomatic or oligosymptomatic CF disease can now be clearly discerned. The clinically relevant intermediate range of 5% to 30% residual activity of CFTR is amenable to semiquantitative diagnostic assessment of CFTR activity. We have learnt over the years that these individuals with inconclusive sweat test and CFTR genetics exhibit individual signatures in sweat test, NPD and ICM. In other words, whereas the within-group variability of NPD and ICM profiles is low in PI CF and non-CF healthy controls, variability is high within the intermediate group of PS CF/CFTR-RD. Virtually each examined subject who had been referred to us to make a diagnosis exhibited a personal fingerprint in his NPD and ICM tracings.
The majority of patients whom we diagnosed with CF or CFTR-RD by extended electrophysiology are carriers of the wild-type CFTR coding sequence on at least one of their CF alleles. The disease-causing genetic lesions should reside in the non-coding region of CFTR or elsewhere in the genome affecting the regulation CFTR expression in a tissue depending fashion31–33 which may explain the large within-group variability of CFTR activity in the respiratory and intestinal epithelium seen in this group.
Acknowledgments
We would like to thank the cystic fibrosis (CF) patients and the healthy volunteers for their participation in nasal potential difference (NPD) and intestinal current measurement (ICM). We are indebted to Hugo de Jonge for the hands-on training with the commercial Ussing chamber equipment and John P Clancy for the instruction into the Cystic Fibrosis Foundation Therapeutics, Inc, Therapeutic Development Network standard operating procedures (SOPs) for NPD measurements. Helpful discussions with Manfred Ballmann, Inez Bronsveld and Nico Derichs during the discussion period for the preparation of the European Cystic Fibrosis Society Diagnostic Network Working Group SOPs for NPD and ICM are gratefully acknowledged. We thank our colleagues at MHH and at the CF centres in Berlin, Bielefeld, Bochum, Bremen, Cologne, Erlangen, Frankfurt, Gießen, Göttingen, Halle, Hamburg, Heidelberg, Kiel, Leipzig, Magdeburg, Münster, Munich, Oldenburg, Regensburg and Vienna for the fruitful cooperation during the set-up of the diagnostic procedures and the diagnostic processing of individual cases.
References
Footnotes
Deceased Died on August 2018
Contributors The nasal potential difference (NPD) and intestinal current measurement (ICM) standard operating procedure protocols were installed on-site by RM, AS, AvB and BT. NPD and ICM measurements were performed and evaluated by RM, AS, NA, AvB, LG, FS, CS, ST and BT. Biopsies were collected by SJ, CM, CD and AMD. CD, GH, SJ, FCR, AS-H, TW, AMD and BT have been involved in the recruitment and discussion of diagnostic cases and the provision of clinical care during follow-up. Data interpretation was performed by BT. The initial draft of the manuscript was written by BT, with all co-authors (except AvB) contributing revisions and all approved the final version.
Funding The Deutsche Forschungsgemeinschaft (grant no SFB 621-C7), the Christiane Herzog Stiftung and the Wilhelm-Hirte-Stiftung provided funds to purchase the equipment to perform nasal potential difference (NPD) and intestinal current measurement (ICM) according to standard operating procedures. Funds for consumables and personnel to run the cystic fibrosis electrophysiology laboratory were provided by the Deutsche Forschungsgemeinschaft (grant no SFB 621-C7; July 2005 to June 2013), the Mukoviszidose eV (grant no: SIP CFTR_3_2010; October 2010 to September 2013) and the German Center for Lung Research at BREATH, Disease Area Cystic Fibrosis (grant no: FZ 82DZL002A1; since January 2013).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval If rectal suction biopsies were taken, prior written consent was obtained from patients and/or their parents. The in-house set-up of the standard operating procedures of the European Cystic Fibrosis Society Diagnostic Network Working Group for intestinal current measurement, including collection of rectal biopsies from healthy non-cystic fibrosis (CF) controls and CF patients was approved by the ethics committee of Hannover Medical School by 12 December 2011 (No. 6071).
Provenance and peer review Not commissioned; internally peer-reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.