Article Text

Abstract
Background Cystic fibrosis (CF) is a genetic, multisystemic, progressive and life-shortening disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. Different genotypes have been linked to variations in disease progression among people with CF. The burden of illness (BOI) in children with CF is incompletely characterised, particularly as it relates to CFTR genotypes prior to the availability of the first CFTR modulators. This retrospective, cross-sectional, descriptive study evaluated the BOI in US children with CF <12 years of age prior to the first approval of CFTR modulators.
Methods Data from the US Cystic Fibrosis Foundation Patient Registry from 2011 were used to summarise key patient and disease characteristics using descriptive statistics, overall and grouped by age (0 to <2 years, 2 to <6 years and 6 to <12 years) and genotype (F508del/F508del, F508del/minimal function (MF), MF/MF, gating mutation on ≥1 allele, residual function mutation on ≥1 allele and R117H on ≥1 allele) group.
Results The analysis included 9185 children. Among 6-year-olds to <12-year-olds, mean (SD) per cent predicted FEV1 in 1 s was 92.6% (17.5%). Among all children <12 years of age, the mean (SD) all-cause hospitalisation and pulmonary exacerbation rates in 2011 were 0.4 (1.0) and 0.3 (0.8), respectively. Most (93.6%) had ≥1 positive lung microbiology culture. CF-related medication and nutritional supplementation use was common across all ages and genotypes. More than half (54.7%) had ≥1 CF-related complication. Evidence of disease burden was observed across the age and genotype groups studied.
Conclusions Prior to the approval of the first CFTR modulator therapies in children <12 years of age, CF was associated with substantial BOI from an early age—including respiratory infections, hospitalisations/pulmonary exacerbations, need for supplemental nutrition and pharmacological treatments—irrespective of genotype.
- cystic fibrosis
Data availability statement
Data sharing is not applicable to this article as no data sets were generated or analysed by the coauthors in the present study. The US CFFPR collects and manages its own data and maintains processes for researchers to request summarised data (https://www.cff.org/Research/Researcher-Resources/Tools-and-Resources/Patient-Registry-Data-Requests/).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What was the burden of illness of cystic fibrosis (CF) in US children <12 years of age prior to the first approval of CF transmembrane conductance regulator (CFTR) modulators?
Prior to CFTR modulator therapies, CF was associated with substantial burden of illness from an early age—including respiratory infections, hospitalisations, pulmonary exacerbations, need for supplemental nutrition and pharmacological treatments—irrespective of genotype.
The results of this retrospective, cross-sectional, descriptive analysis are consistent with the progressive nature of CF from infancy across childhood and demonstrate the substantial disease burden for children with CF, irrespective of genotype.
Introduction
Cystic fibrosis (CF) is a genetic, multisystemic, progressive and life-shortening disease caused by a loss of CF transmembrane conductance regulator (CFTR) function resulting from mutations in the CFTR gene.1 People with CF have mutations in both copies of the CFTR gene, which affects the activity of the CFTR protein. The deficiency in CFTR leads to manifestations that present as early as in utero and progress throughout life.1 2 Prenatally, abnormalities in the digestive, pancreatic and (in males) reproductive systems are observed.1 3 Between infancy and childhood, abnormalities of the pancreas, intestine, liver and airways result in malnutrition, poor growth and lung infections and inflammation.1 2 These manifestations require aggressive symptomatic therapies from an early age.4 5 Symptomatic therapies are essential to preserving lung function and maintaining adequate nutrition and growth.6 7 Inhaled agents, such as dornase alfa and hypertonic saline, and chest physiotherapy are used to improve airway clearance.6 7 Systemic and/or inhaled antibiotics and anti-inflammatory agents are used to treat lung infections.6 Pancreatic enzyme replacement therapy (PERT) and nutritional supplementation are used to maintain adequate nutrition and growth.6 With symptomatic therapies alone, the median predicted survival for people with CF in the USA was 36.8 (95% CI 34.7 to 40.3) years in 2011.8 This increased to 46.2 (95% CI 45.2 to 47.6) years in 2019 with advances in CF care that include the introduction of CFTR modulator therapies, which treat the underlying cause of disease.9 10
More than 2000 CFTR gene mutations have been identified to date.11 The classification of mutations is evolving from a system based on molecular mechanism12 to one based on a combination of molecular mechanism and therapeutic approach/response.7 13 More than 80% of people with CF have at least one F508del-CFTR mutation, and genotypes can be classified based on presence of this common mutation.9 Minimal function mutations are a group of mutations that have no biologic plausibility of translated CFTR protein as predicted by the genetic sequence or for which in vitro testing supports lack of response to tezacaftor, ivacaftor or tezacaftor/ivacaftor and there is evidence of clinical severity on a population basis.14 Gating mutations are defined by a defect in the function of the chloride channel opening and closing, which results in significantly reduced chloride transport.11 12 Residual function mutations are characterised by the presence of chloride transport at the cell surface, although at a reduced level compared with normal.15 16 CFTR genotypes have been linked to variations in the progression of CF,17 and the burden of illness (BOI) in children with CF, particularly as it relates to CFTR genotype groups, is not fully characterised.
The objective of this study was to evaluate the BOI in children <12 years of age with CF in the USA prior to the first approval of CFTR modulators using retrospective, cross-sectional, descriptive analyses.
Methods
The US Cystic Fibrosis Foundation Patient Registry (CFFPR) is an ongoing patient registry first established in the 1960s to collect information about the disease course of CF.18 The registry has excellent coverage of US patients with CF (approximately 84%) and captures patient demographic and clinical characteristics with high accuracy.18 CFFPR data from 2011 were used to describe key patient and disease characteristics in children with CF prior to the first approval of CFTR modulator therapies. In 2011, 27 111 individuals with CF were included in the US CFFPR.8 For inclusion into this study, individuals must have had a CF diagnosis with ≥1 physician visit at an accredited CF centre in 2011 and been <12 years of age as of 1 January 2011. All disease characteristics were reported directly by staff at accredited CF centres through an online data entry system. Disease characteristics queried for this study included pulmonary function and exacerbations, hospitalisations, lung microbiology, CF-related complications and use of nutritional supplementation and CF-related medications.
In children 6 to <12 years of age, lung function was characterised by per cent predicted FEV1 (ppFEV1). Spirometry is not routinely performed in children <6 years of age, and these data are not reported by the US CFFPR.8 The ppFEV1 was calculated using Global Lung Function Initiative 2012 equations,19 which are the current standard and were retroactively applied to 2011 data from CFFPR. The annual ppFEV1 measure for each child was calculated as the average of the child’s single-best measurement from each available quarter. The population mean was calculated as the average of the annual measures.
Mean number of hospitalisations and pulmonary exacerbations (PEx) per child were calculated as the total number of events during 2011 divided by the total number of children. A PEx was defined as a care episode with a ‘pulmonary exacerbation’ reported by the child’s physician.
The percentage of children with positive lung microbiology was calculated as the proportion of children with ≥1 positive culture reported by the centre during 2011. Microorganisms included Staphylococcus aureus, methicillin-resistant (MRSA) and methicillin-sensitive S. aureus, Pseudomonas aeruginosa, Haemophilus influenzae, Burkholderia cepacia, Aspergillus (any species) and nontuberculous mycobacteria.
The percentage of children using CF-related medications was calculated as the proportion of children with a center-reported CF-related prescription during 2011. CF-related medications included antibiotics, antifungals, mucolytics, nonsteroidal anti-inflammatories, hypertonic saline, bronchodilators, corticosteroids, pancreatic enzymes, acid blockers and ursodiol.
Use of nutritional supplementation was calculated as the percentage of children with any centre-reported use during 2011. Types of supplemental feeding mechanisms studied included oral, nasogastric, gastrostomy and jejunal tubes and total parenteral nutritional supplementation.
Centre-reported complications of CF during 2011 were collected through check-all-that-apply fields and included pancreatic, hepatobiliary, pulmonary, gastrointestinal and psychological conditions.
Results were summarised overall and by age and genotype groups using descriptive statistics. Counts and percentages were reported for discrete measures and means, and SDs were reported for continuous measures. Age groups were 0 to <2 years, 2 to <6 years and 6 to <12 years. Genotype groups studied were F508del/F508del, F508del/minimal function (MF), gating mutation on ≥1 allele, residual function (RF) mutation on ≥1 allele, MF/MF and R117H on ≥1 allele. Mutations included in each group are listed in online supplemental table S1. R117H was treated as a separate RF group because the registry had incomplete information on the associated poly-T and TG-tract mutations, which impact the clinical phenotype of these children. These common genotype groups do not encompass all patients with CF and are not all mutually exclusive; thus, the total number of patients included in the genotype analyses does not equal the total patient population in 2011.
Supplemental material
Missing data varied by disease characteristic and were not imputed. The number of evaluable patients with non-missing data is detailed for each characteristic and patient group.
Data availability statement
Data sharing is not applicable to this article as no data sets were generated or analysed by the coauthors in the present study. The US CFFPR collects and manages its own data and maintains processes for researchers to request summarised data (https://www.cff.org/Research/Researcher-Resources/Tools-and-Resources/Patient-Registry-Data-Requests/).
Patient and public involvement
Patients and the public were not involved in the design, conduct, reporting, or dissemination plans of this study.
Results
Study population
A total of 9185 children met the inclusion criteria in 2011, including 4398 (47.9%) children with the F508del/F508del genotype, 1887 (20.5%) with F508del/MF genotypes, 439 (4.8%) with a gating mutation on ≥1 allele, 438 (4.8%) with an RF mutation on ≥1 allele, 336 (3.7%) with MF/MF genotypes and 262 (2.9%) with R117H on ≥1 allele. Sex was balanced overall and in all genotype groups; 49.9% of included children were female. The majority of children were identified as White (92.6%); however, the percentage ranged from 79.5% among MF/MF children to 96.5% among F508del/F508del children (table 1).
Demographics by genotype group*
Pulmonary function, hospitalisations and pulmonary exacerbations
Among all children 6 to <12 years of age, the annual mean (SD) ppFEV1 was 92.6% (17.5%) (table 2), and an estimated 15.8% of children had ppFEV1 <75%. The mean (SD) ppFEV1 was 92.2% (17.5%) among children with the F508del/F508del genotype, 92.1% (17.5%) among children with F508del/MF genotypes, 95.0% (16.6%) among children with a gating mutation on ≥1 allele, 98.3% (16.4%) among children with an RF mutation on ≥1 allele, 87.4% (19.1%) among children with MF/MF genotypes and 97.9% (11.8%) among children with R117H on ≥1 allele (table 2).
ppFEV1 by genotype group in children 6 to <12 years of age*
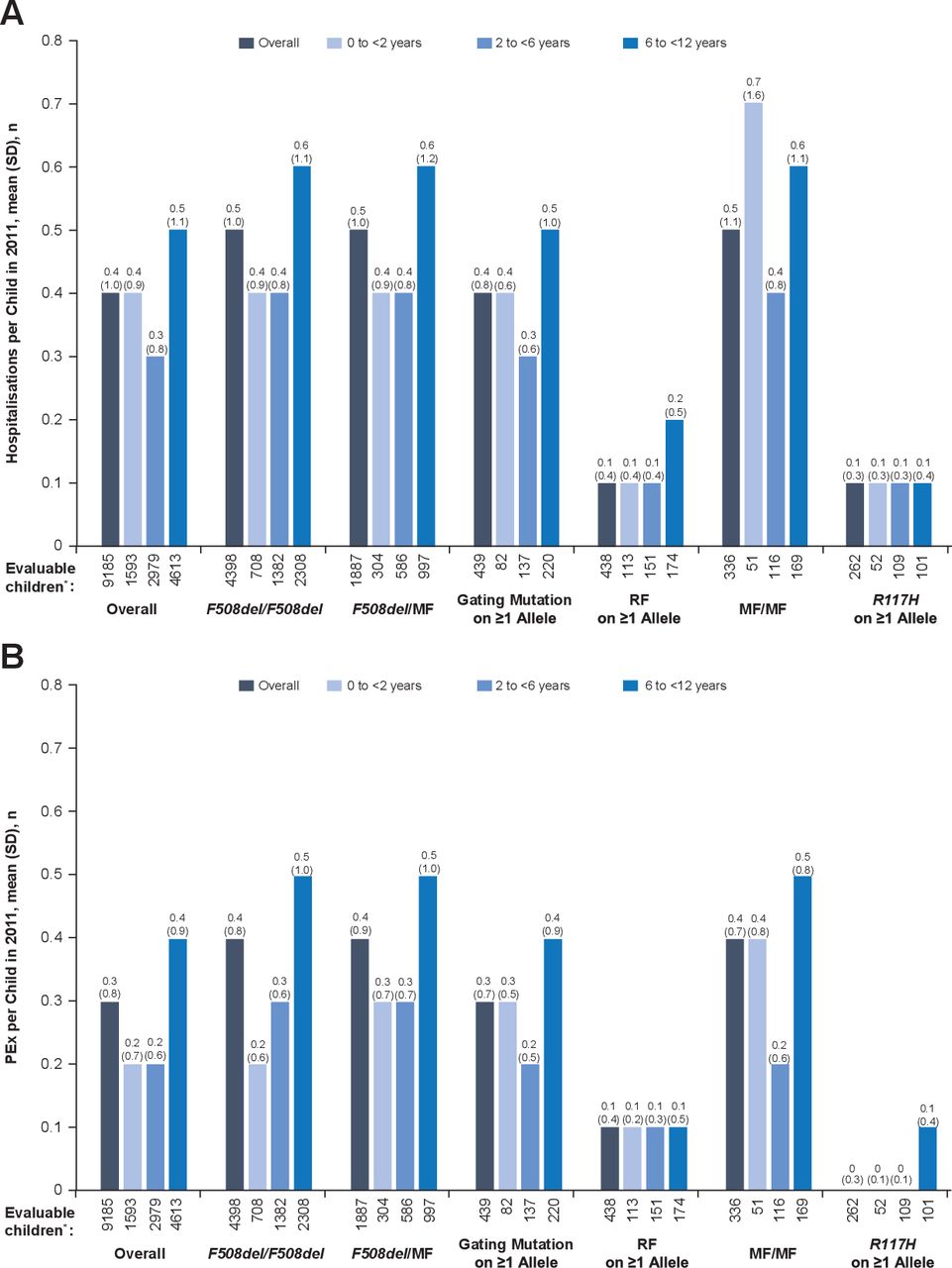
Among all children <12 years of age, the mean (SD) all-cause hospitalisation rate in 2011 was 0.4 (1.0). Annual hospitalisation rates by genotype and age groups are shown in figure 1A. In most genotype groups, the highest hospitalisation rate was seen among children 6 to <12 years of age compared with younger children. Across all age groups, the lowest overall rate of hospitalisation was seen in children with an R117H mutation on ≥1 allele and the highest among children with MF/MF genotypes.
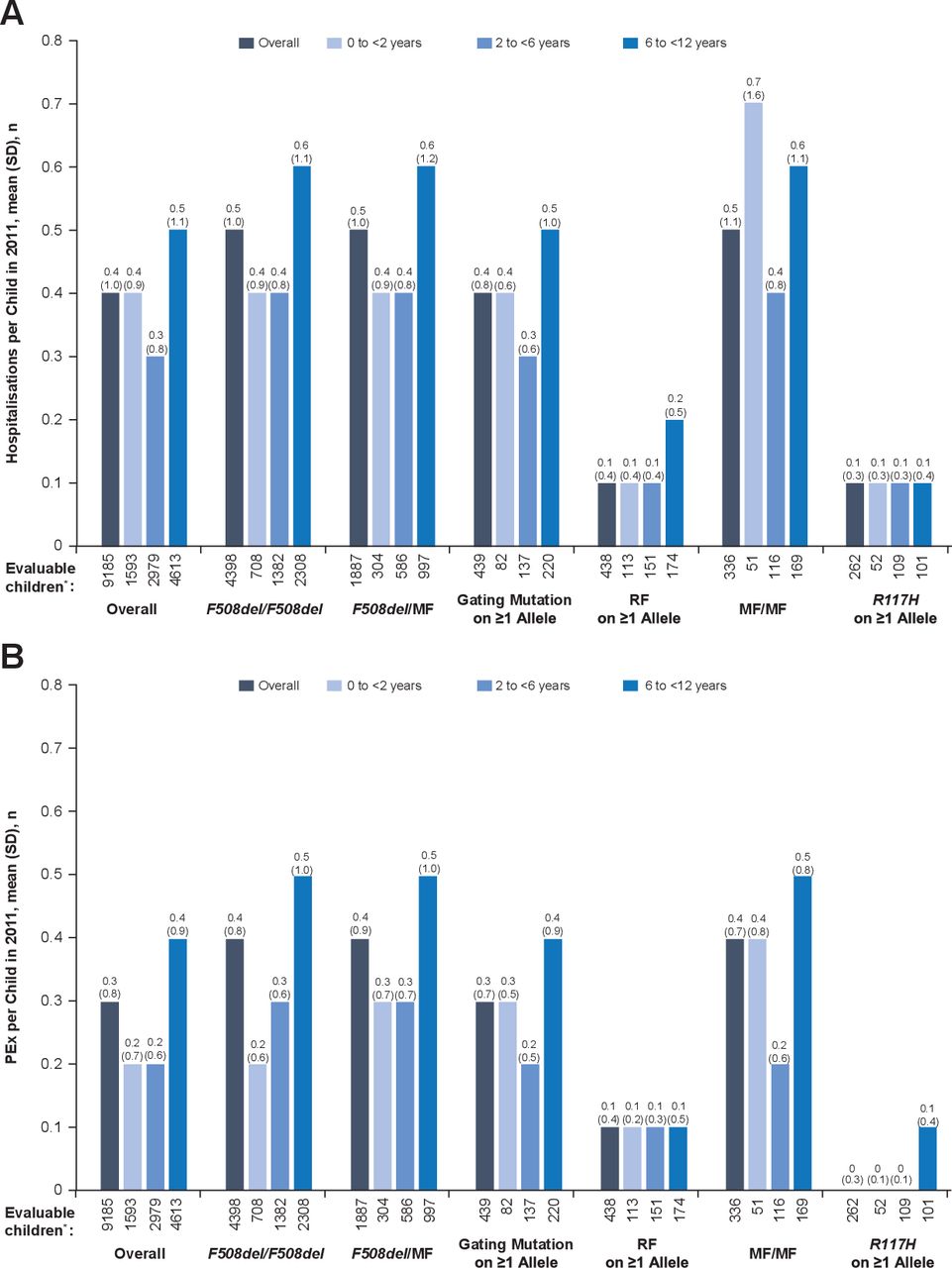
(A) Hospitalisation and (B) PEx rates according to age and genotype group. *‘Evaluable children’ includes all children with non-missing data. MF, minimal function; PEx, pulmonary exacerbation; RF, residual function.
Among all children <12 years of age, the mean (SD) annual rate of PEx was 0.3 (0.8). Annual PEx rates by genotype and age groups are shown in figure 1B. Overall and in most genotype groups, children 6 to <12 years of age had a higher mean number of PEx compared with younger children in 2011. The lowest annual PEx rates were observed among children with RF or R117H mutations on ≥1 allele, and the highest were observed among children with F508del/F508del, F508del/MF, MF/MF genotypes or gating mutations on ≥1 allele.
Lung microbiology
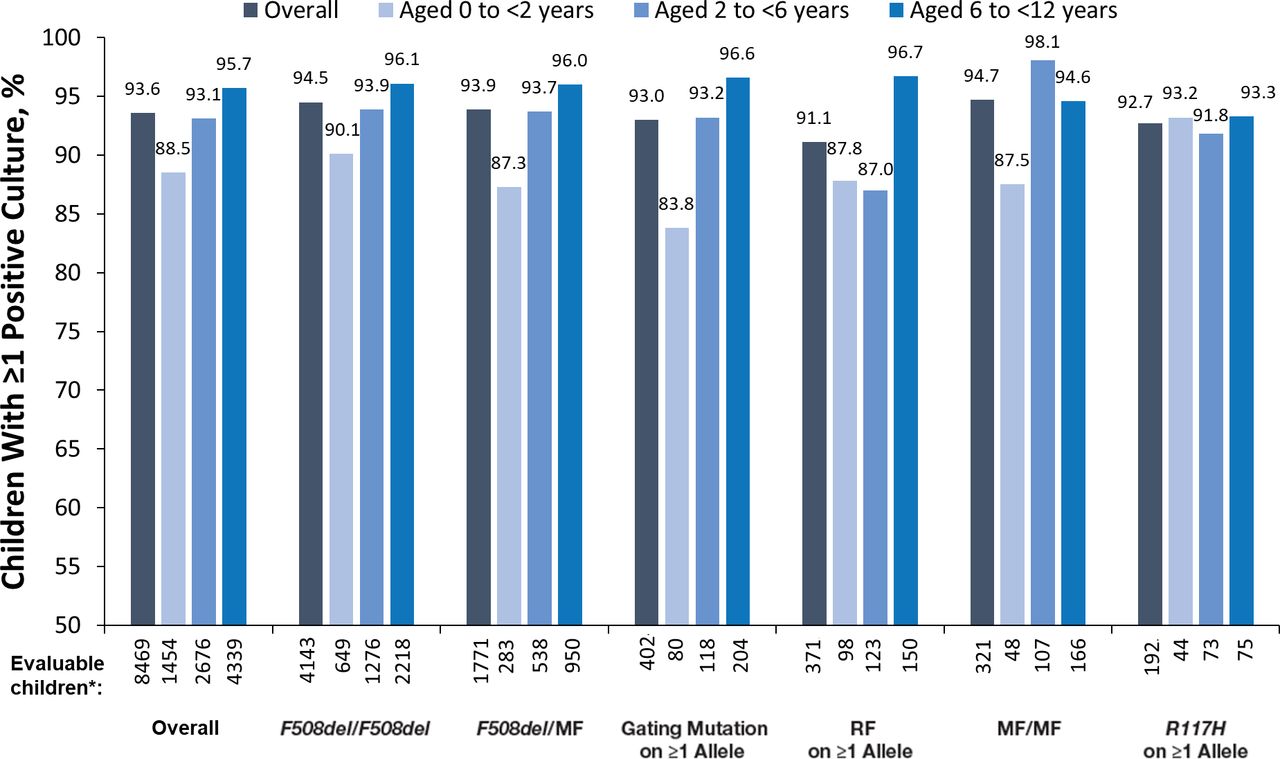
Most (93.6%) children <12 years of age had ≥1 positive lung microbiology culture in 2011. A high proportion of positive microbiology cultures was observed across all age and genotype groups (>80%) (figure 2). In most genotype groups, a higher proportion of positive cultures was observed among children 6 to <12 years compared with children <6 years. This trend was also seen when examining the studied organisms individually, except for H. influenzae, which was less common among the oldest age group. The greatest variation across genotypes in the proportion of positive microbiology was seen in children 6 to <12 years of age for P. aeruginosa (12.2% [R117H on ≥1 allele] to 47.3% [MF/MF]) and MRSA (9.5% [R117H on ≥1 allele] to 33.2% [F508del/MF]; online supplemental figure S1).

Positive lung microbiology rates by age and genotype groups for any positive microbiology. *‘Evaluable children’ includes all children with non-missing data. MF, minimal function; RF, residual function.
Medication use
Use of CF-related medications was common across all age and genotype groups, and use increased across the increasing age groups. The most-used medications among children <12 years of age were inhaled bronchodilators (89.7%), followed by pancreatic enzyme replacement therapy (PERT) (89.3%), dornase alfa (74.0%) and hypertonic saline (41.6%) (online supplemental table S2).
Prevalence of use of inhaled airway clearance medications overall and by age group was similar among children with F508del/F508del, F508del/MF, MF/MF genotypes and gating mutation on ≥1 allele and was lower among children with R117H or RF mutations on ≥1 allele. Similarly, use of PERT varied by genotype group; the vast majority of children with F508del/F508del (99.6%), F508del/MF (98.1%) and MF/MF (97.3%) genotypes or a gating mutation on ≥1 allele (93.6%) used PERT from infancy, while fewer than half of the children with RF mutations (37.2%) or R117H (19.3%) on ≥1 allele used PERT (online supplemental table S2).
Nutritional supplementation
The majority (59%) of children received oral nutritional supplementation. While no difference in the prevalence of oral nutritional supplementation was clearly evident across age groups, differences were observed across genotype groups: oral nutritional supplementation was more common among F508del/F508del, F508del/MF and MF/MF genotype groups and among children with a gating mutation on ≥1 allele, and it was less common among children with RF mutations or R117H on ≥1 allele (online supplemental figure S2A).
Supplemental feeding via a gastronomy tube was received by 13.5% of all children (online supplemental figure S2B). Overall, the observed proportion of children receiving such supplemental feeding was 15.9% among children 6 to <12 years of age, 12.3% among children 2 to <6 years of age and 8.7% among children 0 to <2 years of age. The numerical differences between age groups were driven by the F508del/F508del and F508del/MF groups; other groups did not show obvious differences by age group.
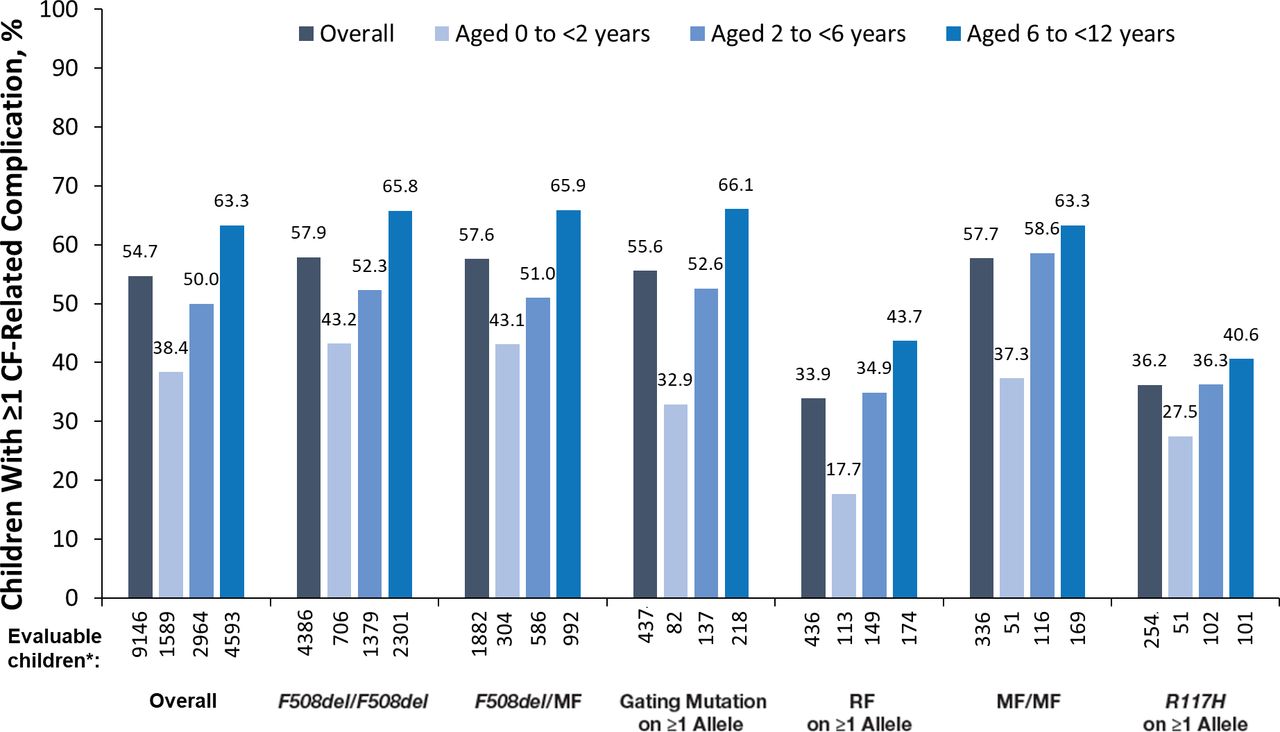
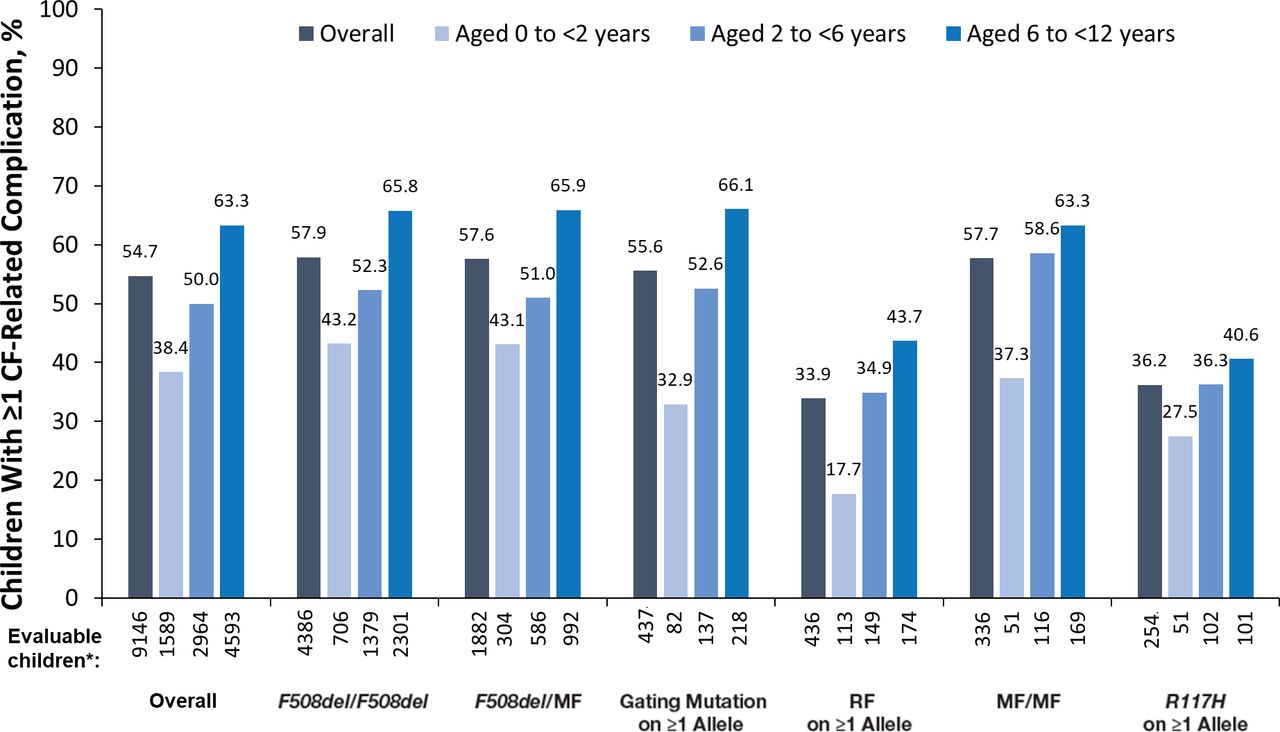
Complications of CF
More than half (54.7%) of all children with CF <12 years of age had ≥1 complication of CF as of 2011. Overall, and within each genotype group, the proportions of children with any complication increased in older age groups (figure 3). Not all complications of CF, however, followed this pattern. Gastro-oesophageal reflux disease (GORD), which was the most common CF-related complication (occurring in 25.7% of all children), was reported among 28.3% of children 0 to <2 years of age, 26.1% of children 2 to <6 years of age and 24.5% of children 6 to <12 years of age. This decrease in the rate of GORD in older age groups was only seen in the F508del/F508del and F508del/MF groups (online supplemental table S3). The second and third most common complications of CF, asthma (22.2% of all children) and sinus disease (15.8% of all children), were reported in higher proportions of children in older age groups, both overall and within the genotype groups (online supplemental table S3). All other complications of CF studied (pancreatic, hepatobiliary, psychological, and additional pulmonary or gastrointestinal) were reported in <15% of the children.
{kind=link}
{kind=link}
{kind=link}
CF-related complications according to age and genotype group. *‘Evaluable children’ includes all children with non-missing data. CF, cystic fibrosis; MF, minimal function; RF, residual function.
Discussion
The results from this analysis illustrate the substantial burden of CF from infancy through childhood and across genotypes prior to the first approval of CFTR modulator therapies. Overall and within most genotype groups, PEx, complications of CF, respiratory infections and need for symptomatic treatments increased as patients aged. More importantly, BOI starts early, as substantiated by the use of symptomatic treatments and supplemental nutrition and by the proportion of children experiencing respiratory infections and complications of CF. These findings highlight the considerable burden of CF from infancy. Lung disease was an important contributor to the burden of CF from an early age and prior to routine assessment of lung function starting at 6 years of age. This was illustrated by the high use of pulmonary-related symptomatic therapies and the high rate of positive lung microbiology cultures, including P. aeruginosa, which is associated with a worse prognosis20 21 despite the use of symptomatic therapies. Of note, the decline with age in the rates of H. influenzae is consistent with the recommended vaccination schedule in the USA.22 Further burden resulted from PEx, which are severe life events associated with high morbidity that can lead to an increased rate of lung function decline, worse long-term prognosis and death.23–29 While reduced lung function, as measured by spirometry (ie, ppFEV1), was observed among children 6 to <12 years of age, the impact of CF on lung function in children <6 years of age is not generally assessed as spirometry is an effort-dependent manoeuvre and may be unreliable or infeasible in this population. Importantly, spirometry is known to have limited sensitivity in evaluating early lung disease; other modalities, such as multiple breath washout (ie, lung clearance index), have demonstrated functional lung disease in those with normal ppFEV1.8 30 These observations underscore the need for more sensitive and age-appropriate methods to assess lung disease in children.31
Additional burden was observed as a result of the multisystemic nature of the disease. The majority (59%) of children received oral nutritional supplementation, with 13.5% requiring gastrostomy tube feeding. The impact of impaired exocrine pancreatic function was illustrated by the high prevalence of PERT use starting in infancy (86.3%). The observation that most children required nutritional supplementation, PERT and other symptomatic therapies suggests that substantial and progressive disease burden persisted and highlights the need for novel therapies that treat the underlying cause of CF.11 32 Early treatment with such therapies provides the potential to slow disease progression and ameliorate the increasing symptomatic treatment and disease burden on patients and their caregivers.11 32
This registry-based descriptive study has inherent limitations. Data were collected by CF centres and submitted to the US CFFPR; thus, reported data may be incomplete or reflect individual centre practices or interpretations. An evaluation of 2012 US CFFPR data suggested that 95% of clinic visits and 90% of hospitalisations were entered into the registry, and an audit of select data fields found high concordance between the medical records and the registry, ranging from 82.6% (aztreonam, tobramycin use) to 99.9% (patient sex).18 Furthermore, this study only included children in the USA, and results may not be generalisable to children with CF outside the USA. Finally, stratification by both age and genotype group led to small numbers for certain subgroups, limiting interpretation.
This study demonstrated that, prior to the first approval of CFTR modulator therapies, CF was associated with substantial BOI from an early age—including respiratory infections, hospitalisations and PEx; need for supplemental nutrition; and high treatment utilisation—irrespective of genotype.
Data availability statement
Data sharing is not applicable to this article as no data sets were generated or analysed by the coauthors in the present study. The US CFFPR collects and manages its own data and maintains processes for researchers to request summarised data (https://www.cff.org/Research/Researcher-Resources/Tools-and-Resources/Patient-Registry-Data-Requests/).
Ethics statements
Patient consent for publication
Acknowledgments
The authors gratefully acknowledge the patients with CF, families and physicians who participated in the Cystic Fibrosis Foundation Patient Registry (CFFPR) and the CFFPR for supplying the data that made this analysis possible. The authors are also grateful to Kelli Edwards-Malone, PharmD (who was an employee of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company), and Francesca Francois, PharmD, MPH (who is an employee of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company), for their coordination and support, along with the medical writing and editorial support by Karen Kaluza Smith, PhD, of ArticulateScience LLC, which received funding from Vertex.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors KB was involved in data collection. KB, EA-S, SJM, GL and CLC contributed to interpretation of the data, drafted and critically revised the manuscript and approved the manuscript for publication. KB accepts full responsibility for the finished work as the guarantor; KB had access to the aggregated data and controlled the decision to publish.
Funding This study was supported by Vertex Pharmaceuticals Incorporated, Boston, MA, USA.
Competing interests KB, EA-S, GL and CLC are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. SJM is an employee of ICON Clinical Research, which received funding from Vertex Pharmaceuticals Incorporated for analytic consulting services for this study and which receives funding from various pharmaceutical, biotechnology and device companies for providing clinical research services.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.