Article Text

Abstract
Introduction Exercise is important in the postoperative management of lung cancer, yet no strong evidence exists for delivery of home-based programmes. Our feasibility (phase I) study established feasibility of a home-based exercise and self-management programme (the programme) delivered postoperatively. This efficacy (phase II) study aims to determine whether the programme, compared with usual care, is effective in improving physical function (primary outcome) in patients after lung cancer surgery.
Methods and analysis This will be a prospective, multisite, two-arm parallel 1:1, randomised controlled superiority trial with assessors blinded to group allocation. 112 participants scheduled for surgery for lung cancer will be recruited and randomised to usual care (no exercise programme) or, usual care plus the 12-week programme. The primary outcome is physical function measured with the EORTC QLQ c30 questionnaire. Secondary outcomes include health-related quality of life (HRQoL); exercise capacity; muscle strength; physical activity levels and patient reported outcomes. HRQoL and patient-reported outcomes will be measured to 12 months, and survival to 5 years. In a substudy, patient experience interviews will be conducted in a subgroup of intervention participants.
Ethics and dissemination Ethics approval was gained from all sites. Results will be submitted for publications in peer-reviewed journals.
Trial registration number ACTRN12617001283369.
- exercise
- lung cancer
- thoracic surgery
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
This trial is testing a novel postoperative 12-week home-based exercise and self-management programme for patients after surgery for lung cancer.
It is a prospective, multisite, two-arm parallel 1:1, randomised controlled superiority trial with assessors blinded to group allocation.
The trial will provide important information related to the effectiveness of this intervention to improve several patient-important outcomes.
Given the inclusion criteria is limited to English speaking adults, expected to be alive greater than 6 months, with a performance status 0–2, and not currently meeting physical activity aerobic guidelines, our trial findings may not be generalisable to all patients undergoing surgery for lung cancer.
Introduction
Lung cancer is associated with significant morbidity and mortality.1 Surgery offers the best chance of survival for patients with early-stage lung cancer, with 5-year survival rates at approximately 60% for patients with localised disease.2 However, physical and psychological impairments after diagnosis are persistent and associated with poor health-related quality of life (HRQoL).3–6 Both global HRQoL and physical function are predictive of survival in lung cancer.7
Patients with lung cancer are a vulnerable group going into surgery; at time of diagnosis they are less physically active and have poorer exercise capacity than similar aged peers without lung cancer.5 8 Over the first 6 months from surgery, functional decline is common, with a spiral of causative factors including symptoms, treatment side effects and physical inactivity.5 8 9 Functional decline in lung cancer is often characterised by worsening physical function, deconditioning, reduced exercise capacity and muscle strength, and physical inactivity.5 Six months after primary cancer treatment, only 30% of patients meet the physical activity recommendations.5
Exercise training is beneficial for patients after surgery for lung cancer.10 A Cochrane review concluded that post-operative exercise training is associated with improved exercise capacity and muscle strength (moderate-certainty evidence), improved physical aspects of HRQoL (low certainty-evidence) and reduced dyspnoea (very low-certainty evidence).11 Six of the eight included trials tested centre-based exercise programmes (hospital or community), however centre-based exercise programmes specific to lung cancer are rare in clinical practice. This is partly due to hospital-related barriers to delivery of such programmes including lack of funding, staffing and infrastructure12 13; patient-related barriers to access programmes including travel time and cost14; and lack of cost-effectiveness data.10 15 None of the trials were purely home-based.11 Home-based exercise programmes for post-operative patients are an alternative model, although evidence is lacking.10 Home programmes are appealing in that they may be less resource intensive (although cost-effectiveness data of home programmes are lacking)10 15; they reduce the requirement for the patient to travel and allow flexibility for them to exercise within their own daily schedule14; and it supports the desire to shift more healthcare away from the hospital with potential health policy implications. Yet they may introduce new barriers such as a requirement for greater motivation from the individual to exercise on their own. Nonetheless, it is a concept worth investigating for the lung cancer field since the evidence consistently shows that exercise is associated with improvement in important patient outcomes, but patients rarely access programmes in clinical practice.10
For these reasons, we developed and subsequently established the feasibility of a new postoperative 12-week home-based exercise and self-management programme (the programme) for patients with lung cancer.16 The programme included behavioural change techniques with the view to maximise adherence of the prescribed exercises and promote prolonged physical activity behavioural change given the programme was conducted in an unsupervised home setting.16 The aim of this efficacy (phase II) trial is now to determine whether the programme is effective in improving physical function (primary outcome) in patients after surgery for lung cancer. If shown to be effective at improving patient outcomes, the next step in this programme will be to explore if home-based exercise can achieve equivalent outcomes to centre-based exercise programmes, as has been demonstrated in the chronic obstructive pulmonary disease field.17
The primary hypothesis of this trial is that the programme compared with usual care will improve physical function 12 weeks after surgery. Secondary hypotheses are that the programme will improve (1) physical function at 6 and 12 months; (2) HRQoL at 3, 6 and 12 months; (3) functional exercise capacity; muscle strength, mass and quality; physical activity levels; and patient reported outcomes at 3 and 6 months; and (4) result in lower healthcare resource usage over 12 months after surgery.
The trial will also include a qualitative sub-study which aims to characterise the views and experiences of patients participating in the programme; as well as a long-term follow-up investigation exploring survival rates to 5 years.
Methods and analysis
Design
This will be a prospective, assessor-blinded multisite, parallel-group, two-arm, randomised controlled superiority trial with a 1:1 allocation ratio to either a 12-week programme of home-based exercise and self-management plus usual care (‘the programme’) or usual care alone. The design is shown in figure 1. The protocol will follow the recommendations of Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT),18 19 Guidelines for Reporting Trial Protocols and Completed Trials Modified Due to the COVID-19 Pandemic and Other Extenuating Circumstances (CONSERVE-SPIRIT extension)20 and Template for Intervention Description and Replication (TIDieR).21

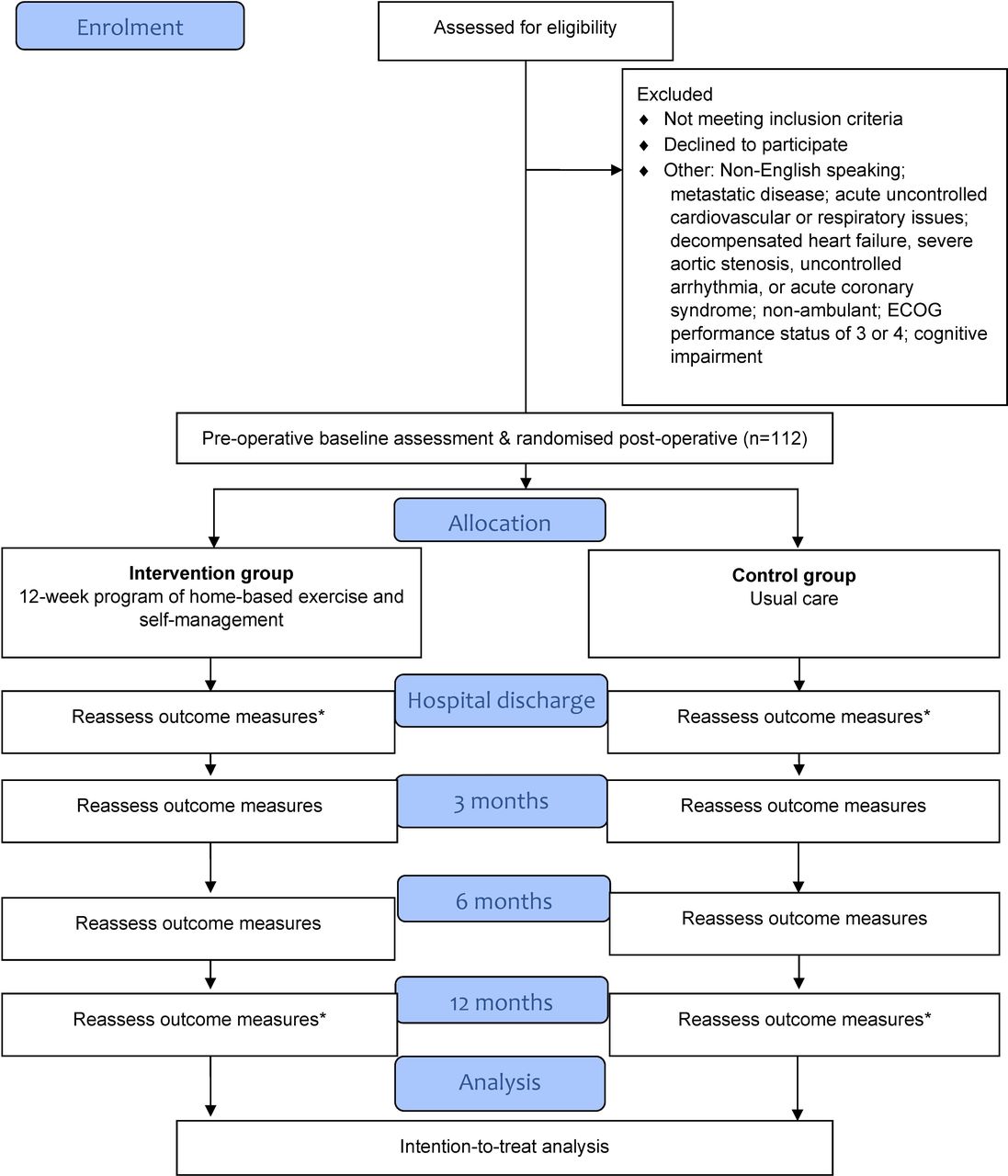
CONSORT flow chart. *Denotes not all outcomes measured at this time point. CONSORT, Consolidated Standards of Reporting Trials; ECOG, Eastern Cooperative Oncology Group.
Participants
Patients will be eligible if they are planned to receive surgical treatment for suspected or confirmed non-small cell lung cancer (NSCLC) and meet the inclusion criteria (table 1). Participants will be provided with written and verbal information; and written informed consent will be obtained prior to enrolment.
Inclusion and exclusion criteria
Recruitment
Participants will be recruited through the lung cancer services at three tertiary hospitals in Melbourne, Australia. Participants will be screened for eligibility prior to enrolment. Enrolment into the trial and subsequent baseline measurement will occur before surgery. The schedule of enrolment is shown in figure 2.

{kind=link}
{kind=link}
Schedule of enrolment, intervention and assessment. *Denotes measure to be completed in a subset of participants only. HRQoL, health-related quality of life.
Randomisation and blinding
The randomisation schedule will be computer generated by an independent statistician and allocation will be carried out centrally by the lead investigator or site trial coordinator using the Research Electronic Data Capture (REDCap) randomisation module. Block permuted randomisation with stratification by hospital site will be used to randomise eligible patients to either intervention or control. Allocation concealment will be achieved as the randomisation code will not be released until the participant has completed the baseline outcome measures and undergone surgery. Randomisation will occur day 1 or 2 postsurgery to ensure all patients have undergone surgery and received confirmation of a diagnosis of NSCLC. If the histological diagnosis is still unconfirmed at this point, the patient will be randomised, retained in the trial and the final diagnoses will be reported at the end of the trial. Following randomisation, the intervention physiotherapist will be contacted by the lead investigator or trial coordinators and informed of the group allocation. Blinding of participants and the intervention physiotherapists cannot be achieved due to the nature of the exercise intervention. Assessors will be blinded to group allocation. If an assessor becomes unblinded, this will be documented and reported. All usual care staff who provide usual clinical care to patients on the ward will be blinded to group allocation. Other study investigators (excluding those involved in delivering the intervention), including the study statistician, will remain blinded until the database has been cleaned, a blinded data review has taken place and the data are ready for analysis.
Usual care: intervention and control groups
Usual care (medical, nursing and allied healthcare including physiotherapy) will be provided in the perioperative period to participants in both intervention and control groups. Outpatient physiotherapy, prehabilitation or exercise programmes will not be provided before or after surgery, as per usual care. Before surgery, patients will receive a hospital-specific information booklet on thoracic surgery, containing information about the surgery and hospital stay, instructions about breathing and coughing, chest and shoulder exercises and advice on the importance of being physically active. After surgery, inpatient physiotherapy will focus on achieving early mobility milestones for discharge home and breathing exercises if required.9 Inpatient physiotherapists providing usual care will be blinded and their inpatient ward treatment(s) will be recorded on the participant’s hospital medical record.
Exercise training and self-management programme: intervention group
In addition to usual care, participants in the intervention group will receive the programme which will commence after surgery. The programme was tested in our feasibility study16 and adjustments for this current efficacy (phase II) trial were made based on the results. These changes and rationale for them are discussed in the feasibility (phase I) publication.16
The programme will include an initial individual face-to-face consultation with the trial intervention physiotherapist (the physiotherapist) on the ward on the day of hospital discharge or the day prior; and then individual weekly telehealth consultations (delivered via telephone) with the physiotherapist to 12 weeks after surgery (table 2). The initial consultation can be split over two sessions if needed, depending on the fatigue and pain levels of the participant after surgery. This will run for 15–45 min in duration. The subsequent follow-up telehealth consultations will be 5–20 min depending on the needs of the participant including their readiness and understanding for behavioural change. Session duration will be recorded.
Intervention components of the 12-week programme
During the initial consultation, the physiotherapist will prescribe and teach the participant an aerobic and resistance exercise programme to perform unsupervised in their home. The exercises are based on physical activity guidelines for people with cancer22 but the specific exercise prescription will be individualised based on each participant’s current ability, symptom levels and goals.23 Participants will also be given an activity tracker to wear daily over the 12 weeks. Table 2 provides details of the programme components. During the weekly telehealth consultations, exercises will be progressed/regressed as needed; and participants will be asked to recall their adherence rate to the exercise programme and report their daily step count.
Initial and follow-up consultations will involve behavioural change techniques based on the model by Health Change Associates.24 Using these techniques, the physiotherapist will work with the participant to identify potential barriers and enablers for them to achieve the prescribed exercise programme and brainstorm strategies to overcome these. In the initial consultations this will include a focus on assessing the participant’s readiness for behavioural change, and their goals and confidence regarding physical activity and working with them (and their carer if the participant desires) to set personalised and individual goals regarding their participation in physical activity. The follow-up consultations will re-visit the participant’s goals, promote adherence, and discuss any new barriers or issues. Details of all consultations will be recorded in standardised online forms for each participant within the REDCap database.
The physiotherapists delivering the intervention will be Australian Health Practitioner Regulation Agency (AHPRA) registered practitioners. The physiotherapists leading the intervention will also undergo training in behavioural change techniques with Health Change Associates.24 All physiotherapists will undergo standardised in-person training in the intervention protocol. They will be given access to audio-recordings of example consultations and detailed instruction manuals. Random audits of audio recorded telehealth consultations and online forms will be performed against the intervention protocol by the lead investigators.
Adherence to the intervention and completion rates will be recorded. The percentage of consultations delivered over the 12 weeks (number delivered against target of 12 sessions—adherence rate) and percentage of participants who continue consultations to 12 weeks (completion rate) will be reported.
Outcome measures
The schedule of outcome measurement is shown in figure 2. Blinded outcome assessors will be AHPRA registered physiotherapists trained in-person in the assessment protocol by investigator SMP. This investigator will also conduct in-person competency checks with the assessors to ensure protocol compliance over time. Assessors will be provided with videos of several of the tests and an instruction manual outlining all instructions.
All assessments will be conducted at the hospital site or in the participant’s home, depending on the participant’s preference and COVID-19 restrictions, except the 12-month assessments which are only phone based (figure 2). In the case that an in-person follow-up assessment is unable to proceed due to COVID-19 restrictions, the assessment will be delayed up to 14 days or converted to a phone-based assessment for questionnaire measures only. Every effort will be made to minimise missing data related to COVID-19 restrictions, and missing data including reasons will be reported at conclusion of the trial.
The primary outcome of physical function will be measured at baseline then 3 (primary endpoint), 6 and 12 months after surgery.25 The European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ c30) was chosen as the primary outcome measure as this is the most commonly used measure in cancer core outcome sets,26 being commonly used to measure HRQoL in lung cancer and general oncology exercise trials.27 28 The physical function domain has strong test retest reliability (r=0.91),29 is sensitive to change,5 25 and was successfully used with participants in our feasibility study.16 The EORTC QLQ c30 is a 30-item self-administered questionnaire that measures HRQoL over the past week. It includes five functional scales (physical, role, emotional, cognitive and social functioning), three symptom scales (pain, fatigue and nausea/vomiting), a global health status/quality of life scale and six single-item scales (appetite loss, dyspnoea, diarrhoea, constipation, insomnia and financial impact).25 The lung cancer (LC13) supplementary module will also be used, which contains items measuring lung cancer specific symptoms and side-effects.30 Responses to most questions are rated on a Likert scale according to occurrence in the past week. All domain and single-item raw scores will be linearly transformed to a scale of 0–100.31 Higher scores on the functional domains and global health status/quality of life scale indicate higher functioning and higher HRQoL.25 31 Lower scores on symptom domains and single items indicate fewer symptoms.25 31 The threshold for clinical importance of the physical function domain is <83/100 points.32
Table 3 describes the secondary outcome measures for the trial. Demographic, medical and social data will be obtained including age, sex, ECOG performance status, cancer stage and type, comorbidities, body mass index, smoking history, respiratory function, education, occupational status, living arrangements, surgical details, hospital length of stay and any postoperative treatment. Survival will be followed to 12 months and 5 years.
Secondary outcomes measures
Participant experience feedback (intervention arm subgroup)
A subgroup of participants in the intervention group will be invited to participate in an individual semistructured interview to record their views and experiences, including behavioural barriers for adherence, of the programme. Purposive sampling will be used to ensure a rich spread of experiences from participants of different ages and residential location (metropolitan/regional). The interview guide will be underpinned by the application of the Theoretical Domains Framework (TDF) and Theoretical Framework of Acceptability (TFA) facilitating in-depth exploration of the behavioural change and intervention acceptability.33 34 The interviews will be conducted using videoconferencing, in-person or via the telephone depending on participant preference. Interviews will be conducted until data saturation is reached, based on interim coding by investigators (SMP and GAW-W) commencing after completion of the first eight interviews. Interviews will be recorded and transcribed verbatim. This sub-study will be reported according to the Consolidated criteria for Reporting Qualitative research guidelines.35
Data management and analysis
All participants will be allocated a confidential trial code. The master coding database will be kept in REDCap with access restricted to the lead investigator and trial site coordinators. All deidentified data will be entered and stored in REDCap. REDCap enables validation rules to be used for data entry, audit trails to track usage and direct export to statistical software packages. Data entry will be performed by trained investigators or research assistants and data checks will be performed regularly by lead investigators. Deidentified paper files will be stored in locked filing cabinets at recruitment sites, accessible only to the research team. Videorecordings and audiorecordings of interviews, interview transcripts, ultrasound images and Actical physical activity data will be deidentified and saved in password protected computer drives only accessible to relevant members of the research team. Interview transcripts will be uploaded to NVivo V.1.0 software (QSR International, 2020). Only summary data, without participant identifying information will be presented or published.
Participants can choose to withdraw from the study or not complete a certain test at any stage. If a participant withdraws, consent will be requested to keep their collected data until that point for analyses, and if they decline this will be removed. If a participant chooses not to perform a test or tests at a certain time point, every effort will be made to collect at least the primary outcome.
Intention-to-treat analyses will be performed including all randomised participants in their allocated group arm. A constrained longitudinal data analysis model will be used to analyse the primary outcome (EORTC QLQ c30) across all time points (baseline, 3, 6 and 12 months after surgery), with study group, time point and a study group by time point interaction and recruitment site included in the model. The model will be restricted to have a common baseline mean score across the study groups based on the assumption that there are no differences in the mean outcome between groups at baseline due to randomisation. The absolute difference in mean change in EORTC QLQ c30 physical function domain from baseline between groups will be estimated (including two-sided 95% CI) at 3 months after surgery (primary time point). The constrained longitudinal data analysis model provides valid inference if the missing data mechanism is at most missing at random. Similar analyses will be conducted for the secondary outcomes. The complier average causal effect will be estimated in secondary analyses, in addition to the intention-to-treat effect, using collected adherence data. Heterogeneity of the intervention effect according to postoperative cancer treatment (no postoperative treatment/postoperative chemotherapy, radiotherapy or chemoradiotherapy) will be assessed in exploratory analyses by including interactions between postoperative treatment and study group. The number and percentage of participants with adverse events will be summarised by study group. A health economic analysis will be run alongside the clinical trial.
Qualitative data will be analysed using directed content analysis. Braun and Clarke’s36 six steps of reflexive thematic analysis will be used to find and organise data into codes informed by the TDF and TFA33 34 by two independent researchers.
Sample size calculation
A total of 56 participants is required in each study group (112 in total) to detect a clinically meaningful difference of 12.9 points in mean EORTC QLQ c30 (primary outcome) at 3-month follow-up (primary time point) between the intervention and control groups (which falls in the range of ‘small’ effect size).37 This assumes 80% power, a two-tailed significance level of 5%, an equal SD of 21.3 in the groups (based on prior published data),16 no correlation between baseline and 3 months (conservative) and 20% attrition.5 38
Safety and adverse event reporting
Adverse events will be recorded in REDCap database and reported to the lead investigator. Serious adverse events are defined as any events which are life-threatening or result in death or hospitalisation (or prolongation of hospitalisation), incapacity or disability. Serious adverse events will be reported to the ethics committee within 24 hours of occurrence. Minor adverse events include new or progressive pain, non-injurious falls, severe dyspnoea, worsening fatigue, palpitations, neurological deficits and altered cognitive status. Intervention participants will be asked during telehealth consultations to report any adverse events.
Trial committees
The lead investigator (CLG) will meet with the site trial coordinators at least twice per month during active recruitment and follow-up phases. The lead investigators (CLG, SMP, LD and LE) will meet at least quarterly during the study period. Annual reporting will be provided to funder and ethics committees.
Dissemination
The trial will be reported according to Consolidated Standards of Reporting Trials Statement for Randomised Trials of Nonpharmacologic Treatments.39 Main findings will be submitted to a peer-reviewed journal for publication. Additional papers are expected related to the qualitative substudy, 5-year survival and in-depth exploration of muscle ultrasound. Results will be presented at conferences, consumer forums and through social media to disseminate with the public.
Trial status and summary
This is an Australian-based randomised trial comparing a 12-week programme of home-based exercise and self-management, to usual care, to improve physical function in patients after surgery for lung cancer. The current protocol is number 1 V.4.0 dated 13 May 2021. The trial was registered and approved on the Australian and New Zealand Clinical Trials Registry prospectively in September 2017 and last updated 5 October 2021. Recruitment commenced at site 1 on 23 November 2017, at site 2 in September 2021 and is expected to commence at site 3 in early 2022. Main data collection for the trial (excluding survival data) is expected to be completed by June 2023. Five-year survival data are expected to be collected by mid-2027.
CONSERVE-SPIRIT extension statement
The COVID-19 pandemic has led to a small number of trial modifications20 noting that the trial was designed and commenced prior to the pandemic and is planned to continue throughout the pandemic. Important modifications were made in 2020 (online supplemental appendix 1) these were: update of the protocol (and ethics amendment) to allow participants an option to conduct interviews via videoconferencing; ability to complete only the questionnaire portion of the assessment at follow-up when restrictions prevent in-person assessments; extension of planned recruitment time frames to allow for pauses in recruitment due to government and/or local hospital COVID-19 restrictions and delayed opening of the trial at two sites; and extension of funding to accommodate extended time frames. The modified timeline expects the trial to be completed in June 2023. Modifications were planned by the lead investigators and reviewed and approved by the ethics committee and funding bodies.
Supplemental material
Supplemental material
Ethics statements
Patient consent for publication
Ethics approval
The trial was approved by Melbourne Health Human Research Ethics Committee (HREC/17/MH/182). All protocol changes will be submitted for ethics approval and amended on the registry. Study will be conducted according to the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice (ICH-GCP) guidelines.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
LD and SMP are joint senior authors.
Contributors CLG, LD, SMP and LE designed the trial protocol. CLG procured funding for the trial with assistance from SMP, LD, LI, GAW-W, CFM and MK. CLG, PA, GAW-W, CFM and GW contributed to ethics/governance applications at their hospitals. CLG, SMP and LE drafted the first version of the manuscript. KEL contributed to the design, randomisation, data management and analysis, and sample size sections of the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by Victorian Cancer Agency Clinical Research Fellowship (grant number CRF16011), Cancer Council Victoria (Grant-In-Aid) and The University of Melbourne (Melbourne Clinical and Translational Sciences research platform seed grant).
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.