Article Text

Abstract
Background The diagnosis of idiopathic pulmonary fibrosis (IPF) is often delayed up to several years. The objective of this study was to assess the impact of the diagnostic delay on progression-free survival, quality of life and hospitalisation rates.
Methods A total of 264 incident patients with IPF were included immediately after their diagnosis and followed for up to 5 years, with regular collection of clinical data, quality-of-life questionnaires and assessment of disease progression. Hospitalisation data were extracted from electronic patient records. Analyses were performed on the entire cohort and strata according to forced vital capacity (FVC) at diagnosis.
Results A long diagnostic delay (>1 year) was associated with worse progression-free survival compared with a short diagnostic delay (<1 year) (HR: 1.70, 95% CI: 1.18 to 2.46, p=0.004) especially in patients with mild disease at the time of diagnosis (FVC>80% predicted). Mean total scores of the St. George’s respiratory questionnaire (SGRQ), a derived IPF-specific version of the SGRQ and the chronic obstructive pulmonary disease assessment test (CAT) were consistently higher in patients with long diagnostic delays, indicating worse quality of life. Mean hospitalisation rates were higher during the first year after diagnosis (Incidence rate ratio [IRR]: 3.28, 95% CI: 1.35 to 8.55, p=0.01) and during the entire follow-up (IRR: 1.74, 95% CI: 1.01 to 3.02, p=0.04).
Conclusion A diagnostic delay of more than 1 year negatively impacts progression-free survival, quality of life and hospitalisation rates in patients with IPF. These findings highlight the importance of an early diagnosis for proper management of IPF.
Trial registration number NCT02755441.
- Interstitial Fibrosis
- Rare lung diseases
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Due to national law, sharing data on individuals is not allowed.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The diagnosis of idiopathic pulmonary fibrosis (IPF) is often delayed for up to several years. Consequently, the initiation of effective antifibrotic treatment is also delayed. The impact of this delay in the era of effective antifibrotic treatment is largely unknown.
WHAT THIS STUDY ADDS
In a large and well-characterised cohort of incident patients with IPF, a longer diagnostic delay was associated with worse progression-free survival and quality of life after diagnosis, even after rigorously adjusting for possible bias and confounding. A longer delay was also associated with a higher rate of hospital admissions after diagnosis.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Using real-world data, we highlight the importance and potential benefit of an early diagnosis in IPF patients. An early diagnosis and initiation of antifibrotic treatment could help with the dismal prognosis of IPF.
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive fibrotic lung disease with a dismal prognosis.1 Patient surveys and registry-based studies have shown a significant diagnostic delay, that is, the time from onset of symptoms until a final diagnosis is made, with a median length reported to be between 0.6 and 2.3 years.2–8
Data on the consequences of a diagnostic delay are scarce and, in some cases, conflicting. One early report showed worse survival in patients with a long delay of referral to a specialist centre, while another showed worse survival in patients consulting within 6 months after the beginning of symptoms.2 9 However, these data were collected before approval of antifibrotic treatment and can be difficult to distinguish from lead time bias.2 9 A recent and larger cohort study has not been able to confirm a survival benefit of a short diagnostic delay.10
In the era of effective antifibrotic treatment, there are multiple potential benefits of a shortened diagnostic delay. Patients with early disease show the same rate of disease progression as patients with more advanced disease, supporting early initiation of antifibrotic treatment.11 In addition, consistent data indicate high efficacy of antifibrotic treatment also in patients with less advanced disease.12–14 Finally, antifibrotic treatment can slow disease progression but cannot reverse the established fibrosis in IPF. Consequently, lung tissue lost due to fibrosis, and the resulting accelerated loss in pulmonary function, is not regained when antifibrotic treatment is delayed.15
A long diagnostic delay could impact antifibrotic treatment, disease course and quality of life after diagnosis. Quality of life is impaired in patients with IPF already during the early stages, with a significant deterioration in the last 6 months before death.16 An early diagnosis, relevant treatment initiation and avoidance of ineffective treatments before symptoms increase could potentially alter the disease trajectory and preserve quality of life for a longer period.
The objective of this study was to investigate the impact of diagnostic delay in a well-characterised cohort of incident IPF patients by assessing disease progression, survival, quality of life and hospitalisation rates.
Methods
Patient cohort
The Pulmonary Fibrosis Biomarker (PFBIO) cohort is an ongoing cohort of incident patients with a multidisciplinary team diagnosis of IPF according to international guidelines.8 17 Patients are recruited immediately after their diagnosis, before initiation of antifibrotic treatment. The only exclusion criterion is inability to provide written informed consent. Patients have been recruited at the two largest interstitial lung disease (ILD) centres in Denmark (Herlev and Gentofte University Hospital and Aarhus University Hospital) and are followed for up to 5 years with visits at baseline, 6 and 12 months, and annually thereafter. At each visit, blood samples and clinical data (including quality-of-life questionnaires and pulmonary function tests) are collected.
Data on hospitalisations during follow-up were collected from the patients’ electronic records, covering all hospital admissions in Denmark, and classified according to the cause of admission as respiratory or non-respiratory. Data on patients’ vital status were collected from the Danish Civil Registration System, which is updated daily.
Definition of delay
The diagnostic delay was defined as the time between the first occurrence of any IPF-related symptom (online supplemental table E1), as reported by the patient, and the date of the IPF diagnosis. If patients could not recall the precise time point of symptom onset but rather reported a time period, the midpoint of this period was chosen as the starting date of the diagnostic delay.
Supplemental material
Definition of disease severity and progression
Patients with a forced vital capacity (FVC)≤80% predicted were considered as having ‘mild disease’. However, there is no standardised and wide spread staging system for IPF, and patients with relatively preserved FVC can still be symptomatic.18 Disease progression for the Cox proportional hazards analyses was based on clinical data collected at each visit. Progression-free survival was defined as the time until the first of the following events: a relative decline of FVC≥10% predicted from baseline, a relative decline of diffusing capacity for carbon monoxide (DLCO)≥15% predicted from baseline, or death.
Patient-reported outcome measures
Two quality-of-life questionnaires, the St. George’s Respiratory Questionnaire (SGRQ) and the chronic obstructive pulmonary disease (COPD) assessment test (CAT), were completed at each visit. A validated IPF-specific version of the SGRQ (SGRQ-Ider), analogous to the SGRQ-I was derived from the original SGRQ, as previously described.19 20 In addition, two symptom-specific scores for patient-reported dyspnoea (graded from 0 (when I walk up a hill or one flight of stairs I am not breathless) to 5 (when I walk up a hill or one flight of stairs I am very breathless)) and cough (graded from 0 (I never cough) to 5 (I cough all the time)) were extracted from the CAT questionnaire.
Statistical analyses
The diagnostic delay was transformed into a dichotomous variable using a delay of 1 year as cut-off.
Univariate analyses were used to compare baseline characteristics using t-tests or χ2 tests, as appropriate for the data.
Multivariate analyses were adjusted for age, sex, FVC % predicted at baseline, DLCO % predicted at baseline and antifibrotic treatment (pirfenidone, nintedanib or none) received during follow-up. A sensitivity analysis, also including emphysema as covariate, is included in the online supplemental material. FVC was suspected and confirmed to act as an effect modifier of the diagnostic delay in the Cox proportional hazards models. Consequently, these models were stratified according to FVC above or below 80% predicted, allowing different impact of the diagnostic delay in patients with different baseline FVC. Subgroup multivariate analyses were adjusted for the same covariates as above, including FVC as a continuous variable. Further model control did not reveal any additional interactions.
Analysis of repeated measurements of quality-of-life scores was performed by linear mixed effect models due to non-independence of datapoints within subjects. The models used fixed effects for the visit number in addition to the same covariates as the Cox proportional hazards models and a random effect within each subject. Random intercepts but not slopes were allowed for all subjects. The mixed models used an unstructured variance–covariance structure. Marginal means were calculated based on the mixed effect models at baseline, at 6 months and at 12 months.
Analysis of hospitalisation rates was performed by negative binomial regression analysis, due to right skewness and zero inflation of the data. The models were adjusted for the same covariates as the Cox proportional hazards models.
All analyses were performed with R (R Core Team, 2019, V.3.5.1). No missing data were imputed. Two-sided p values below 0.05 were considered statistically significant.
Results
From April 2016 to June 2021, 264 incident patients with IPF were included in the PFBIO cohort at two ILD centres (online supplemental figure E1). The median diagnostic delay (the time from symptom onset until diagnosis) was 2.0 years (IQR: 0.9–5.0). Baseline characteristics of patients with a short diagnostic delay (<1 year) and patients with a long diagnostic delay (>1 year) are presented in table 1. Overall, at the time of diagnosis, patients with a short delay did not differ significantly from patients with a long delay in univariate analysis. The presenting symptoms were similar in the two groups (online supplemental table E1). The frequency of a truly incidental diagnosis, where the patient was completely asymptomatic at the time of diagnosis, was similar in patients with a short or a long diagnostic delay (1.3% and 2.7%, respectively, p=0.48).
Baseline characteristics at the time of diagnosis of patients with a short (<1 year) or long (>1 year) diagnostic delay
In patients, with mild disease at the time of diagnosis (FVC>80% predicted), a long diagnostic delay was associated with lower FVC, higher SGRQ total score, higher SGRQ-Ider total score and higher dyspnoea score (table 2). In patients with moderate-to-severe disease at the time of diagnosis (FVC≤80% predicted), none of these associations were present (table 2).
Baseline mean (SD) physiologic and quality-of-life measurements in four subgroups based on disease severity at the time of diagnosis and the length of total diagnostic delay
Disease progression and survival
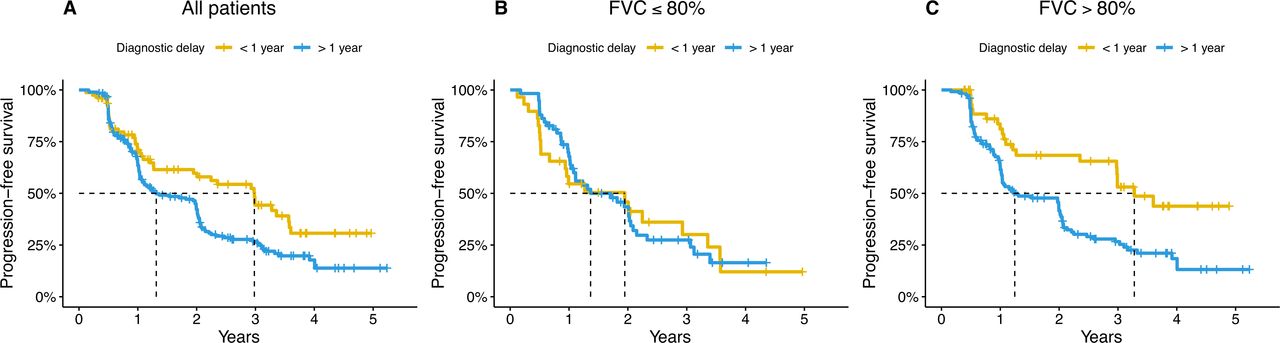
A long diagnostic delay of more than 1 year was associated with a worse progression-free survival compared with patients with a short delay (HR: 1.70, 95% CI: 1.18 to 2.46, p=0.004) in multivariate analysis (figure 1). Patients with a long delay had a median time to progression or death of 15 months compared with 36 months for patients with a short delay (figure 1). However, the diagnostic delay was not significantly associated with all-cause survival alone, not counting disease progression as an event (HR: 1.54, 95% CI: 0.95 to 2.51, p=0.08) (online supplemental figure E2).
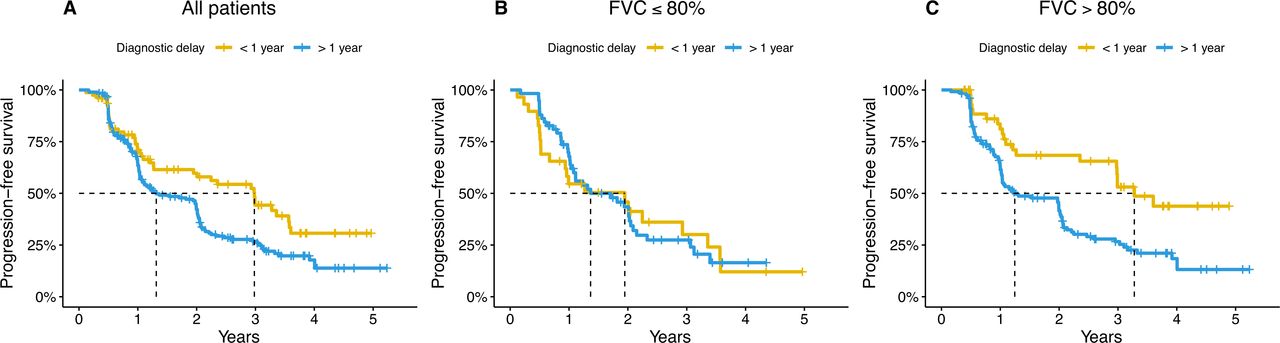
Progression-free survival in patients with a short (<1 year) or long (>1 year) diagnostic delay in the entire cohort (A) and stratified according to forced vital capacity (FVC) at the time of diagnosis≤80% predicted (B) or >80% predicted (C).
The effect of the diagnostic delay on progression-free survival was modified by disease severity at baseline, with an interaction term between FVC and diagnostic delay being significant at p=0.01 in the multivariate Cox proportional hazards model. We therefore conducted stratified analysis in two strata: FVC above and below 80% predicted at the time of diagnosis (figure 1).
A long diagnostic delay was associated with worse progression-free survival in patients with mild disease (FVC>80% predicted) (HR: 2.43, 95% CI: 1.45 to 4.01, p<0.001) but not in patients moderate-to-severe disease (FVC≤80% predicted) at the time of diagnosis (HR: 0.91, 95% CI: 0.51 to 1.62, p=0.76) (figure 1). A sensitivity analysis, also adjusting for lung emphysema, showed similar results (online supplemental tables E2 and E6).
Quality of life
In patients with mild disease at the time of diagnosis, total scores of the SGRQ and SGRQ-Ider were higher (indicating lower quality of life) in patients with a long delay (table 2). This association was not present in patients with moderate-to-severe disease at the time of diagnosis.
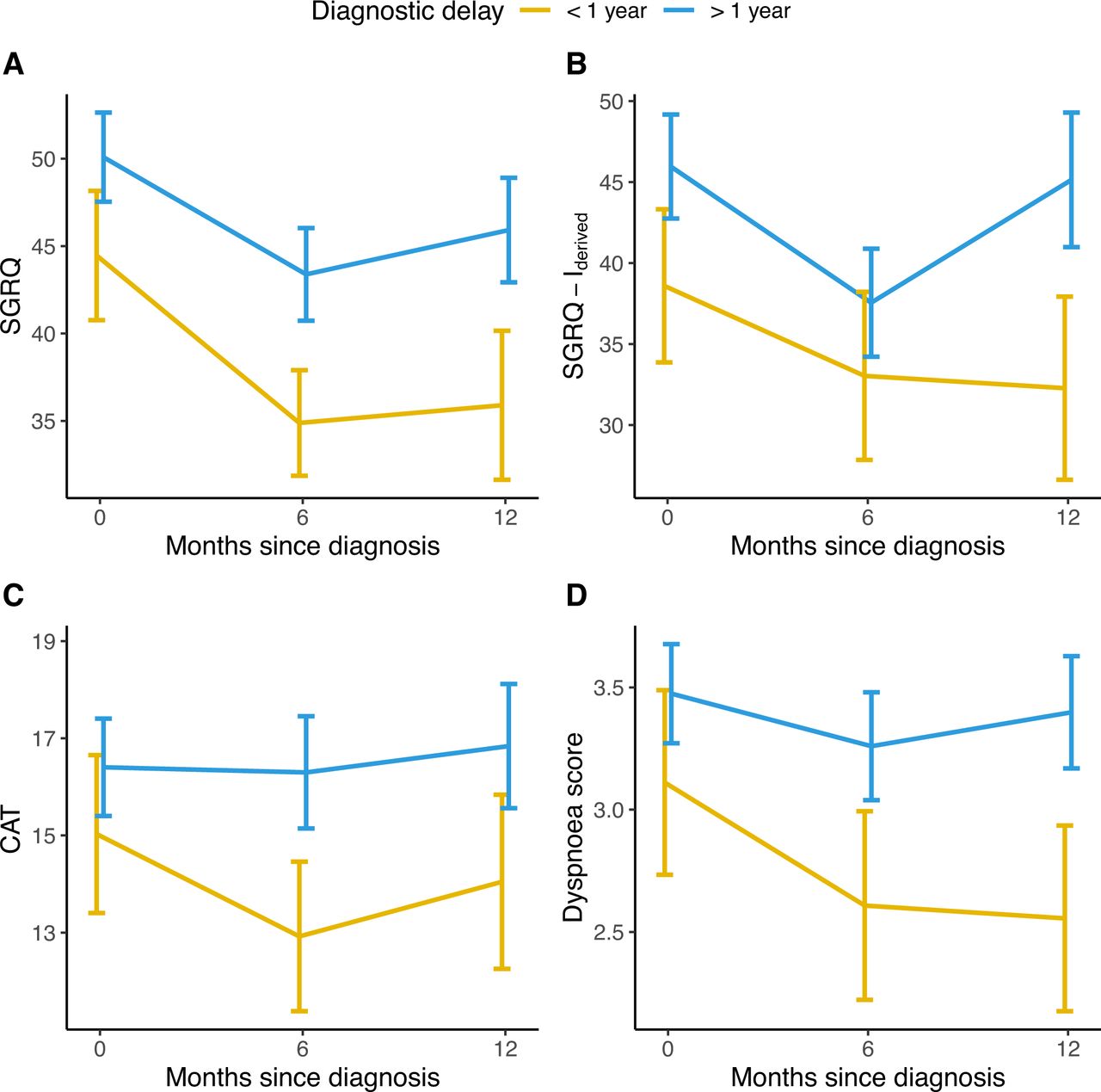
For longitudinal analysis, we used multivariate linear mixed models to compare patients with a long delay with patients with a short delay. A long diagnostic delay was associated with higher marginal means of SGRQ total score (at baseline 50.9 vs 45.2, at 6 months 46.3 vs 40.6 and at 12 months 48.5 vs 42.8; p value for the fixed effect of diagnostic delay=0.04), SGRQ-Ider total score (at baseline 45.8 vs 38.7, at 6 months 40.3 vs 33.2 and at 12 months 47.0 vs 39.9; p=0.04) and CAT total score (at baseline 17.1 vs 14.9, at 6 months 16.8 vs 14.7 and at 12 months 17.9 vs 15.7; p=0.02) (figure 2). In subgroup analyses, these results were consistent in patients with mild disease but not in patients with moderate-to-severe disease at the time of diagnosis (online supplemental figures E3 and E4).
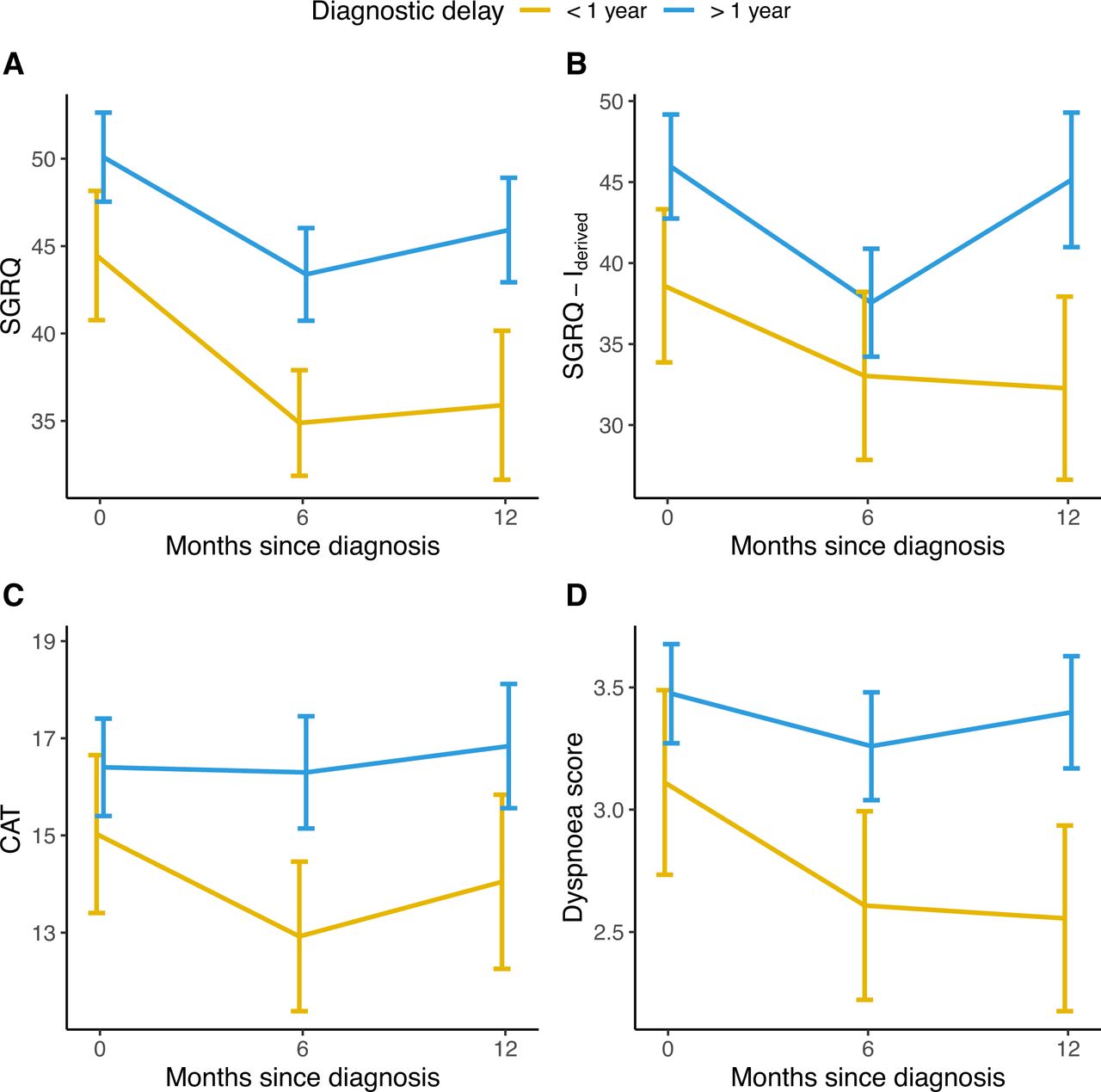
Unadjusted mean total scores of the SGRQ (A), IPF-specific SGRQ-I derived from the original SGRQ (B), CAT (C) and mean individual score from the CAT questionnaire covering dyspnoea (D) during the first year of follow-up in patients with a short (<1 year) or long (>1 year) diagnostic delay. Higher scores indicate worse quality of life for SGRQ, SGRQ-Ider and CAT. Error bars indicate 95% CIs. CAT, chronic obstructive pulmonary disease (COPD) assessment test; SGRQ, St. George’s Respiratory Questionnaire; SGRQ-Ider, idiopathic pulmonary fibrosis (IPF)-specific SGRQ, derived from the original SGRQ.
A patient reported dyspnoea score, derived from the CAT questionnaire, was consistently higher in patients with a long diagnostic delay (at baseline 3.63 vs 3.08, at 6 months 3.44 vs 2.89 and at 12 months 3.62 vs 3.07; p=0.002) (figure 2) while no significant difference was found for the CAT cough score (at baseline 2.43 vs 2.35, at 6 months 2.17 vs 2.10 and at 12 months 2.38 vs 2.30; p=0.63).
Hospitalisation rates
Patients with a long diagnostic delay had higher crude hospitalisation rates after diagnosis compared with patients with a short delay (0.68 vs 0.57 all cause admissions and 0.35 vs 0.27 respiratory admissions per person-year at risk). During the first year after diagnosis, this difference was even more pronounced (0.71 vs 0.38 all cause admissions and 0.25 vs 0.13 respiratory admissions per person-year at risk) (figure 3).

{kind=link}
{kind=link}
{kind=link}
Mean all cause hospitalisation and respiratory hospitalisation rates in patients with a short (<1 year) or long (>1 year) diagnostic delay. Error bars indicate 95% CIs.
In multivariate negative binomial regression analysis, patients with a long delay had higher all cause admission rates during the first year after diagnosis (Incidence rate ratio [IRR]: 3.28, 95% CI: 1.35 to 8.55, p=0.01) and during the entire follow-up period (IRR: 1.74, 95% CI: 1.01 to 3.02, p=0.04) (figure 3). Respiratory admission rates were likewise increased during the first year after diagnosis (IRR: 5.80, 95% CI: 1.19 to 42.12, p=0.04) but not the entire follow-up period (IRR: 1.70, 95% CI: 0.81 to 3.65, p=0.16) (figure 3). In subgroup analyses, these results were consistent in patients with mild disease but not in patients with moderate-to-severe disease at the time of diagnosis (online supplemental figure E5).
Discussion
We have demonstrated a clinically relevant negative impact of a diagnostic delay on progression-free survival, quality of life and hospitalisation rates in a large real-life and well-characterised cohort of patients with IPF.
We found a robust association between a long diagnostic delay and worse progression-free survival in patients with newly diagnosed IPF after adjusting for potential confounding variables. Assessment of the impact of the diagnostic delay is complicated by the different clinical presentations that can result in a short delay. Incidental findings leading to a diagnosis of IPF in asymptomatic patients as well as rapidly progressive disease with a high burden of symptoms both can result in a short delay. The latter presentation has been reported in up to 16% of patients diagnosed with IPF.6 9 In an attempt to isolate these patients with rapidly progressive disease, we performed survival analyses in two strata according to baseline FVC. In patients with baseline FVC>80% predicted, that is, mild disease at the time of diagnosis, the length of the diagnostic delay had greater impact on progression-free survival after diagnosis, in contrast to patients with baseline FVC≤80% predicted, that is, moderate-to-severe disease at the time of diagnosis. In patients whose FVC had declined below 80% predicted within 1 year from the onset of symptoms (ie, patients with a diagnostic delay of less than 1 year and an FVC≤80% predicted at the time of diagnosis) the impact of the diagnostic delay on progression free survival was not significant, possibly due to a rapidly progressive disease phenotype with less time to benefit from an earlier diagnosis and initiation of antifibrotic treatment. These findings are in line with a study of a population, with more advanced disease than the PFBIO cohort, where a short time from the onset of symptoms to consultation in these ‘rapid progressors’ was in fact associated with worse survival.9 We suggest that the potential gain from a shortened diagnostic delay could be greatest in patients with relatively preserved FVC (>80% predicted) at the time of diagnosis who nevertheless are known to progress at a similar rate as patients with more advanced disease,11 and thus have the same benefit from antifibrotic treatment.
A valid criticism of our findings could be that an element of lead time bias cannot be entirely ruled out. However, several aspects of our findings argue against the possibility that they merely represent lead time bias. First, disease severity was similar at the time of diagnosis in patients with short and long diagnostic delay. Considering the progressive nature of IPF, significant lead time bias would give the appearance of less advanced disease in the group with a short diagnostic delay. Second, lead time bias would be expected to impact survival, but not the observed change in disease progression, the improved quality of life measures and lower hospitalisation rates in patients with a short delay. Third, adjusting all analyses for disease severity at the time of diagnosis and antifibrotic treatment received during follow-up, further reduces the impact any lead time bias could have on our results.
Our results also suggest that spirometry, if used alone, would not be an optimal screening tool for IPF. First, many patients had normal FVC at the time of diagnosis and would be missed by spirometry screening, as previously reported.21 Second, our results suggest that patients with preserved FVC>80% predicted, which is often used as threshold for an abnormal test, have the greatest benefit from a short diagnostic delay. A relevant screening tool should therefore rather be optimised in identifying patients with early disease, and could involve a combination of respiratory symptoms, chest auscultation, spirometry and CT scan of the chest.
One important limitation of using FVC to stage disease severity is, that it can be affected by other factors, including other chronic lung diseases. Most notably, lung emphysema could mask the fibrosis related decline in FVC, making interpretation difficult.22 Concomitant emphysema could thus be a contributing factor in the subgroup with relatively preserved FVC>80%, which had a wide range of FVC. Patients with combined pulmonary fibrosis and emphysema have been increasingly recognised as a distinct clinical entity.22 Although a sensitivity analysis, adjusting for the presence of emphysema at baseline, did not change our conclusions, we only had dichotomised data about emphysema available. Ongoing research of the PFBIO cohort, quantifying the extent of emphysema in participants, will increase our knowledge of the relationship between pulmonary function tests, emphysema and disease progression.
The diagnostic delay also had a consistent impact on patient quality of life, symptoms and hospitalisation rates after diagnosis. Even after adjusting for potential confounding variables, such as disease severity and antifibrotic treatment given after diagnosis, there remained a consistent association between a long diagnostic delay and worse quality-of-life scores. The magnitude of these effects was larger than the known minimal clinically important differences for the SGRQ and CAT.23 24 This difference in patient quality of life could in part be attributed to an increased perception and different coping with disease symptoms in patients with a prolonged delay, which often is characterised by misdiagnosis and consultation of many physicians.5 6 The diagnostic process itself has been suggested to affect patients’ quality of life.4
We used the lung-specific quality-of-life questionnaires available in the PFBIO cohort, namely the SGRQ and CAT questionnaires. Although the SGRQ has been validated in patients with IPF, these questionnaires are not specifically developed for use in IPF.25 26 More specific questionnaires such as the IPF-specific version of the SGRQ have been created and validated and would be preferred.19 27 28 Consequently, we used an IPF-specific version of the SGRQ derived from the original SGRQ, which confirmed the association between a long diagnostic delay and impaired quality of life.20 Also, the original SGRQ and CAT have both been used in patients with IPF, and have proven to possess validity to differentiate patients who progress over time.29–31
The higher hospitalisation rates in patients with a long diagnostic delay provide an additional and clinically meaningful outcome of increased morbidity in these patients.32 However, it remains to be established whether the increased hospitalisation rates are due to an increased morbidity or altered healthcare seeking behaviour in patients who have experienced a long diagnostic delay.
Strengths and limitations
Our study has several strengths, which support our findings. First, the PFBIO cohort has prospectively included patients with IPF immediately after their diagnosis, minimising recall bias, which allows for a precise estimate of the diagnostic delay. Second, recruitment of incident patients avoids survivor bias, which could be introduced by recruitment of a prevalent patient cohort. Third, patients were followed with extensive data collection for up to 5 years, allowing for a precise estimate of disease progression and quality of life.
Our study also has some limitations. As this is an observational study, we cannot prove a causative effect of the diagnostic delay on the presented outcome measures. However, the consistent impact of a diagnostic delay on several subjective and objective measures, after adjustment for potential confounders, robustness in sensitivity analyses, as well as the temporal sequence favours a true impact of the diagnostic delay.
An additional limitation could be our use of FVC for stratified analysis, due to the variability and generality of FVC, which can be influenced by other factors than IPF. Thus, using it as a measure of IPF severity is not unproblematic.18 However, adjusting analyses for the presence of emphysema, which could mask a decline in FVC, did not alter our results. Nevertheless, some patients with preserved FVC could have more severe disease than the FVC suggests, as can be seen in the impaired DLCO and peripheral capillary oxygen saturation also in this subgroup.
In conclusion, we report a consistent negative impact of a diagnostic delay of more than 1 year on progression-free survival, quality of life and hospitalisation rates in patients with IPF. These findings were most pronounced in patients with mild disease (FVC>80% predicted) at the time of diagnosis highlighting the importance and potential benefit of an early diagnosis for proper management of IPF patients.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Due to national law, sharing data on individuals is not allowed.
Ethics statements
Patient consent for publication
Ethics approval
Patients are included in the PFBIO cohort after providing written informed consent. The cohort is approved by the Danish capital regional ethical committee (H-16001790) and the Danish Data Protection Agency (HGH-2016-017). This study is performed according to the Declaration of Helsinki.
Acknowledgments
The authors whole heartedly thank all participating patients for their generous contribution to the presented work. The authors also thank the ILD nurses Ruth Fuursted, Yvonne P. B. Mackeprang, Pia G. Kjerrumgaard, Gitte H. Clausen, Lena Jakobsen, Ann Ravnsborg, Louise L. Hegnsbøl and Susanne R. Mørch at Herlev and Gentofte Hospital for their great help with the PFBIO cohort.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors NH designed the study, designed data collection tools, monitored data collection, performed the statistical analysis, cleaned and analysed the data, and drafted and revised the paper. NH acts as guarantor for this study. TSP monitored data collection, collected data, critically interpreted the analysis and revised the paper. EB critically interpreted the analysis and revised the paper. SBS designed the study, designed the data collection tools, critically interpreted the analysis and revised the paper.
Funding This study was funded by unrestricted research grants from Aase og Ejnar Danielsens Fond (Aase and Einar Danielsens Foundation), Roche a/s and Skibsreder Per Henriksen, R. og Hustrus Fond (Shipowner Per Henriksen, R and wife Foundation). The funding sources had no influence on the study design; the collection, analysis or interpretation of the data; in the writing of the report; or in the decision to submit the paper for publication.
Competing interests NH reports unrestricted research grants from Aase og Ejnar Danielsens Fond, Roche and Skibsreder Per Henriksen, R. og Hustrus Fond. TSP reports an unrestricted research grant from Boehringer Ingelheim, contact with Galapagos and consulting fees from Boehringer Ingelheim. EB reports payment for presentations from Boehringer Ingelheim, Hofmann la Roche, Galapagos and Astra Zeneca, support for attending meetings from Boehringer Ingelheim and Hofmann la Roche, and participation in advisory boards for Boehringer Ingelheim and Hofmann la Roche. SBS reports payment for presentations from Boehringer Ingelheim and Roche, support for attending meetings from Boehringer Ingelheim, and participation in advisory board for Boehringer Ingelheim.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.