Article Text

Abstract
Asthma is a frequent respiratory condition whose pathophysiology relies on altered interactions between bronchial epithelium, smooth muscle cells (SMC) and immune responses. Those leads to classical hallmarks of asthma: airway hyper-responsiveness, bronchial remodelling and chronic inflammation. Airway smooth muscle biology and pathophysiological implication in asthma are now better understood. Precise deciphering of intracellular signalling pathways regulating smooth muscle contraction highlighted the critical roles played by small GTPases of Rho superfamily. Beyond contractile considerations, active involvement of airway smooth muscle in bronchial remodelling mechanisms is now established. Not only cytokines and growth factors, such as fibroblats growth factor or transforming growth factor-β, but also extracellular matrix composition have been demonstrated as potent phenotype modifiers for airway SMC. Although basic science knowledge has grown significantly, little of it has translated into improvement in asthma clinical practice. Evaluation of airway smooth muscle function is still limited to its contractile activity. Moreover, it relies on tools, such as spirometry, that give only an overall assessment and not a specific one. Interesting technics such as forced oscillometry or specific imagery (CT and MRI) give new perspectives to evaluate other aspects of airway muscle such as bronchial remodelling. Finally, except for the refinement of conventional bronchodilators, no new drug therapy directly targeting airway smooth muscle proved its efficacy. Bronchial thermoplasty is an innovative and efficient therapeutic strategy but is only restricted to a small proportion of severe asthmatic patients. New diagnostic and therapeutic strategies specifically oriented toward airway smooth muscle are needed to improve global asthma care.
- Asthma
- Asthma Mechanisms
- Asthma Pharmacology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Airway smooth muscle cells (aSMC) derive mainly during embryogenesis from mesenchymal precursors, in parallel with epithelial buds, and are associated with the correct development of the airways tree.1 Present all along the respiratory tree from the trachea to the bronchioles, it is conserved in vertebrae through evolution.2 aSMCs are thought to maintain basal tone in bronchi and homogeneous lung ventilation through modulation of local airflow resistance.3 Though, its real physiological role after development remains controversial in part due to the difficulties to design experimental procedure to test specific hypothesis (impact on mucus expulsion and cough, ventilation to perfusion matching etc).4 However, while the physiological role of aSMC is controversial, their involvement in airways diseases, especially in asthma, is now better understood.
Asthma is a respiratory condition defined by the association of variable respiratory symptoms, such as acute dyspnoea, chest tighness, wheezing and chronic cough, associated with impaired airflow and chronic bronchial inflammation.5 It is a frequent disease affecting around 250 million patients worldwide with an increased incidence for the last decade.6 Asthma treatment is mainly based on inhaled corticosteroids associated with long-acting and short-acting bronchodilators. The main objective of those treatments is to achieve a complete control of the disease.7
Asthma pathophysiological hallmark can principally be divided into three inter-related components: airway inflammation, airway hyper-responsiveness and airway remodelling (figure 1).
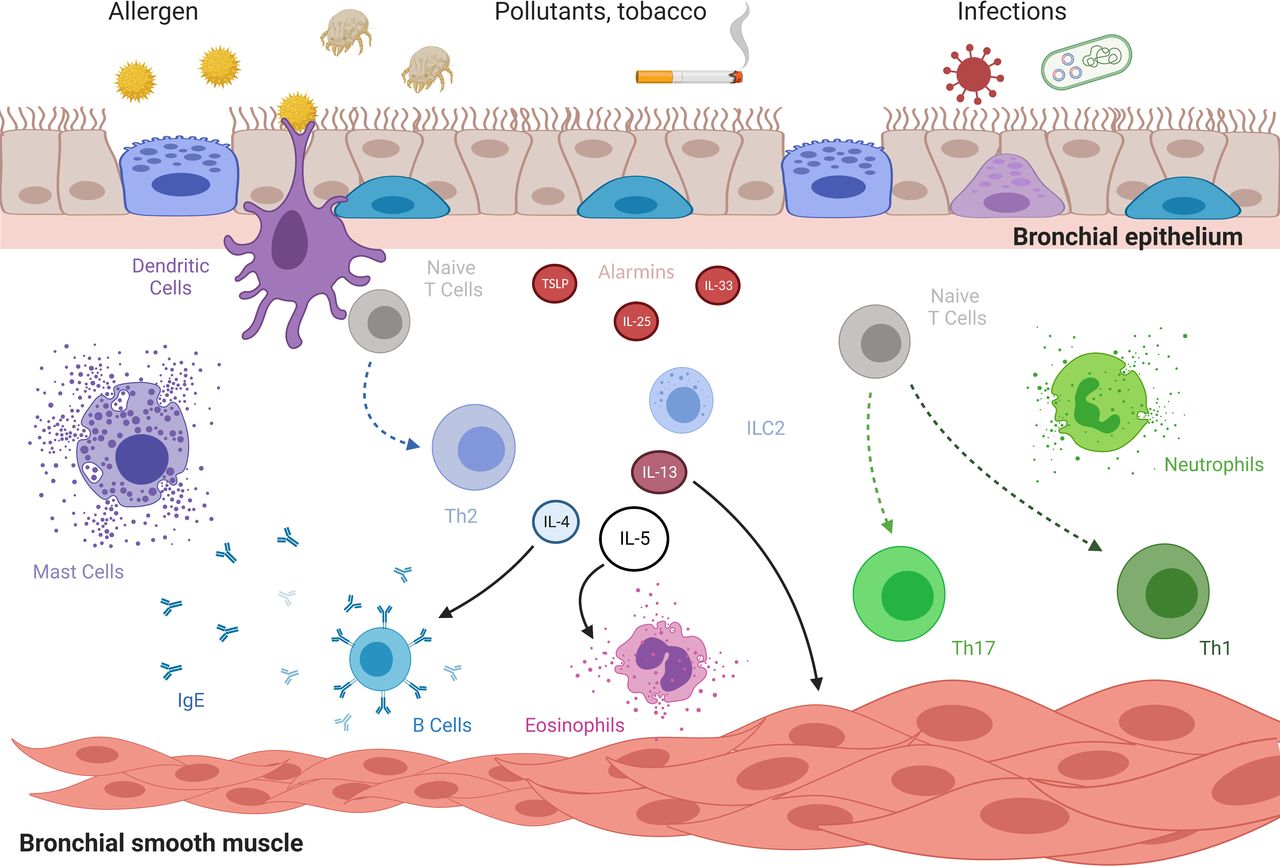
Pathophysiology of asthma. Under stimulation by noxious and/or harmless environment, bronchial epithelium secretes alarmins. Such molecules stimulate innate and adaptative immunity giving rise to infiltration of bronchi by eosinophils or neutrophils or both. Conversely, under sustained inflammation, bronchial remodelling developed with increased basal membrane thickness and hypertrophy and hyperplasy of bronchial smooth muscle. Along with specific bronchial smooth muscle cells acquired hypercontractility, all those mechanisms participate to airway hyper-responsiveness. IgE, immunoglobulin E; IL, interleukine; Th, T-helper cell, TSLP, thymic stromal lymphopoietin. Created with BioRender.com.
Inflammatory pathways involved in asthma course have been extensively studied. Two main pathophysiological pathways are now commonly accepted: eosinophilic (or type-2-driven asthma) and non-eosinophilic.8 Considering type-2-driven asthma, key inflammatory cytokines can be highlighted: interleukine (IL)-4 implicated in T-helper 2 polarisation and IgE switching, IL-5 associated with eosinophils production and trafficking and IL-13 that plays a central role in airway remodelling. Some biotherapies targeting those cytokines pathways had proven their efficiency in selected severe asthmatics and are now available in clinical practice.9 On the contrary, non-eosinophilic asthma, including paucigranulocytic and neutrophilic asthma, remains poorly understood.
Airway remodelling is defined by an association of structural modifications of bronchial wall including aSMC alterations, epithelial dysfunction, reticular membrane thickening and oedema.10 11 Such lesions can be irreversible and lead to the progressive loss of respiratory function through time.12 Airway remodelling unlinked with airway smooth muscle biology is presented elsewhere.13
Airways hyper-responsiveness (AHR) is defined by an exaggerated response of airways to harmless or harmful stimuli. This altered response of bronchi to environment depends on bronchial smooth muscle’s activity, principal actor of bronchoconstriction. Noteworthy, inflammation, by acting on bronchial smooth muscle, is also closely linked to this phenomenon.
Although recent biotherapies critically improved asthma outcomes in severe asthmatic patients, some inflammatory endotypes, noteworthy non-eosinophilic asthma, remain orphan of efficient treatment.14–17 New strategies are evaluated in preclinical setup on non-inflammatory components, notably in aSMC. In this review, we will discuss the biological dysfunctions of the bronchial smooth muscle during asthma, the different techniques of evaluation of these dysfunctions in the clinic as well as the existing or developing therapeutic strategies to manage them.
Biology of aSMC and role in asthma pathophysiology
aSMC contraction dysfunction in asthma
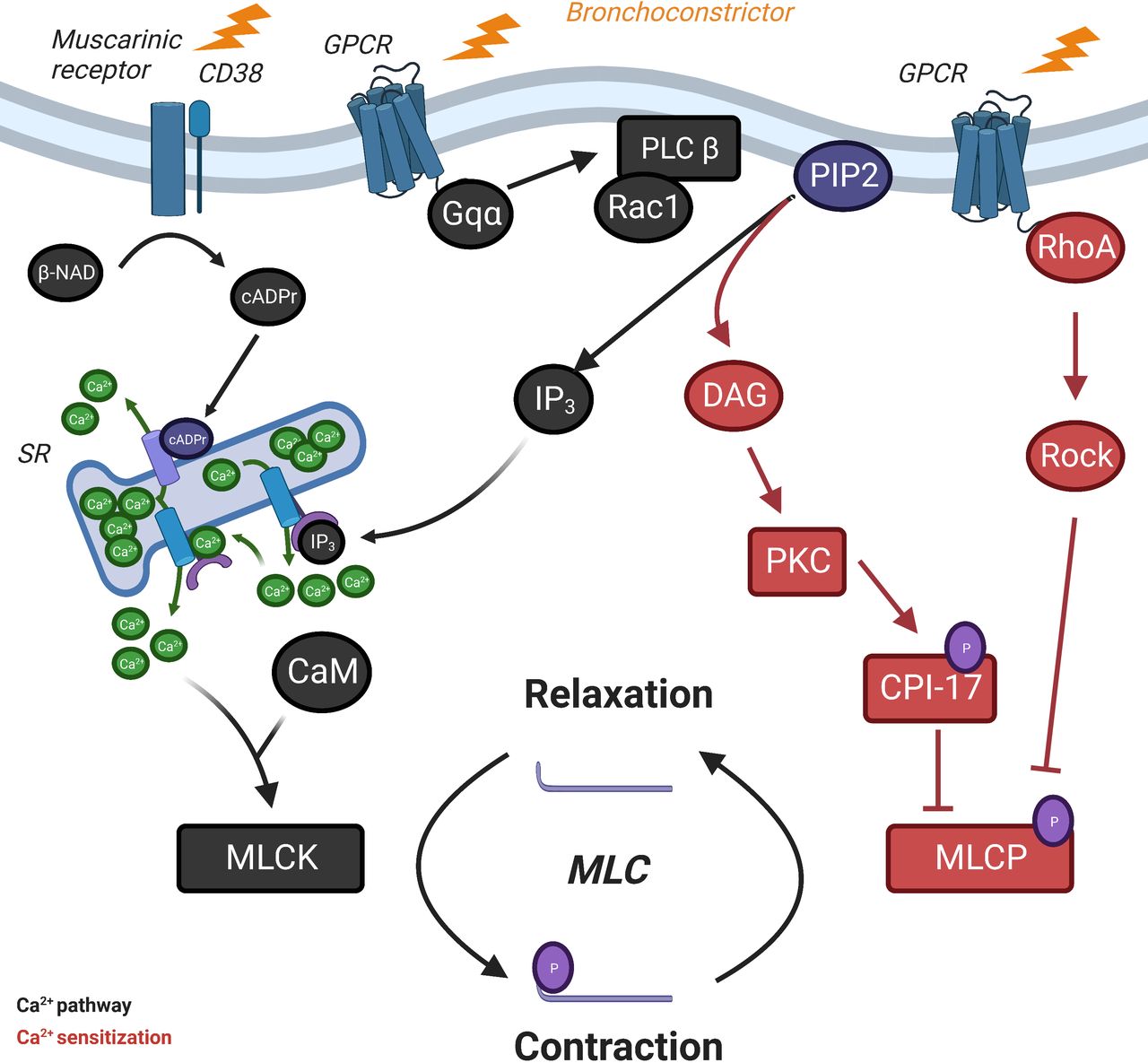
aSMC is essential in the development and sustainment of AHR. Ex vivo studies demonstrated that aSMC harvested from asthmatic subjects displayed increased maximum capacity and shortening velocity compared with controls.18 Moreover, aSMCs isolated from asthmatic patients presented a stronger contraction in response to histamine compared with controls.19 Contractile capacity of bronchial SMC principally depends on the phosphorylation of the 20 kDa myosin light chain (MLC20), which leads to the activation of the contractil apparatus (figure 2). The phosphorylation level of MLC20 is regulated by two enzymes: the myosin light chain kinase (MLCK) and the myosin light chain phosphatase (MLCP). Two distinct signalling pathways regulate the activity of these enzymes: the Ca2+ and the Ca2+ sensitisation pathway.

Intracellular regulation of airway smooth muscle cells contraction. Smooth muscle cells contraction and relaxation cycle depends on phosphorylation and dephosphorylation of MLC, respectively. Increase of intracellular Ca2+ concentration in response to bronchoconstrictor stimuli leads to the CaM-dependent activation of MLCK, which phosphorylates MLC: it is the Ca2+ pathway. In parallel, activation of RhoA–Rock pathways and CPI-17 deactivates MLCP, which prevents MLC from dephosphorylation: it is the Ca2+ sensitisation. β-NAD, beta-nicotinamide adenine dinucleotide; cADP-r, cyclic adenosine diphosphate ribose; CaM; calmodulin; CD, cluster of differentiation; DAG, diacylglycerol; GPCR, G protein-coupled receptors; IP3, inositol triphosphate; MLC, myosin light chain; MLCK, myosin light chain kinase; MLCP, mysoin light chain phosphatase; PKC, protein kinase C; PLC, phospholipase C; PIP2, phosphatidylinositol-4,5-bisphosphate; Rac1, Rac family small GTPase 1; ROCK, Rho kinase; RhoA, Rho family small GTPase A; SR, sarcoplasmic reticulum.
Considering the Ca2+pathway, the phosphorylation of MLC20 by MLCK leads to cross bridges with actin that conducts the aSMC contraction cycle. Interestingly, overexpression of MLCK in aSMC has been observed during asthma associated with its overcontractility.18 20 MLCK activity is principally controlled by the rise in cytosolic Ca2+ concentration coming from extracellular calcium influx through ion channels and sarcoplasmic reticulum (SR) calcium stores.21 SR stores release is principaly triggered by the inositol 1,4,5-trisphosphate (IP3) produced by activated phospholipase C (PLC) after the binding contractile agonists to G protein-coupled receptors or to M3 muscarinic receptors.22
Recently, this signalling pathway has been complemented by studies demonstrating the involvement of the monomeric GTPase Rac1 in aSMC contraction. Rac1 protein is activated in murine and human aSMC by bronchoconstrictors such as methacholine leading to its association with the pleckstrin homology domain of PLC β2 to potentiate the production of IP3 required for aSMC contraction.23 The relevance of this signalling pathway in aSMC was highlighted by demonstrating that Rac1 was overactivated in aSMC from asthmatic patients as well as in aSMC from mice developing allergic asthma. In this experimental model, deletion of the Rac1 gene specifically in SMCs or pharmacological inhibition of Rac1 activity prevents AHR. These results identify the Rac1 protein as a new therapeutic target in respiratory pathologies associated with AHR.23
Independently of IP3 production, cyclic adenosine diphosphate ribose (cADPr) could activate the ryanodine receptors channel (RyR) on the SR leading to the liberation of Ca2+ from the internal SR stores.24 25 Such metabolite is produced from beta-nicotinamide adenine dinucleotide next to the stimulation of the muscarinic receptor M3. Moreover, Ca2+ liberation is increased by Ca2+-induced Ca2+ release through RyRs, resulting in Ca2+ wave propagation and the simultaneous aSMC twitching.26 In asthma, overexpression of CD38 induced by proinflammatory cytokines such as IL-1β, IL-13 and tumor necrosis factor-α leads to increased RyR activation by cADPr.24 27
Calcium uptake and sustainment of ATP synthesis by mitochondria are essential to smooth muscle contraction.28 Calcium uptake is mediated by the mitochondrial Ca2+ uniporter while its release mainly depends on Na2+/Ca2+ or H+/Ca2+ exchanger.29 In aSMC from asthmatic patients, mitochondrial dysfunction can be observed. Downregulation of the expression of sarcoendoplasmic Ca2+ ATPases 2 is associated with the dysregulation of Ca2+ homeostasis in asthma.30
The Ca2+ sensitisation pathway leads to a maximal contraction independently of the intracellular Ca2+ concentration by the regulation of the MLC20 phosphorylation state by MLCP. On the one hand, activation of proteine kinase C by diacylglycerol leads to the phosphorylation of protein kinase C-potentiated phosphatase inhibitor protein of 17 kDa (CPI-17), which binds to the catalytic subunit of MLCP, inhibiting its phosphatase activity.31 On the other hand, activated RhoA interacts with its downstream effector Rho kinase (ROCK), which inactivates MLCP by phosphorylating its myosin-binding subunit.32 Moreover, ROCK can also regulate the activity of MLCP by the phosphorylation of CPI-17.31 33 Interestingly, expression and activity of CPI-17 are increased in aSMC from rats model of allergic asthma.34 In parallel, several studies have shown an increase of RhoA expression in aSMC in animal models.35 Inhibition of RhoA activity prevents and reverses AHR induced in allergic asthma models of guinea pigs.36 Conversely, inflammatory cytokines can significantly influence Ca2+ sensitisation pathway. IL17A, a cytokine secreted by Th17 cells, is able to induce an upregulation of RhoA protein in human aSMC.37 Conversely, it can also increase aSMC contraction by the activation of RhoA–ROCK2 through NF-κB pathway.38 Targeting IL-17 pathway with neutralising antibody decreased the expression of NF-κB, ROCK-I and ROCK-II in lung parenchyma in a mouse model of asthma compared with controls.39 Interestingly, it has been recently proven in mouse that combination of anti-IL17 antibodies along with ROCK inhibitor (Y-27632) significantly improved respiratory resistance, bronchial remodelling and inflammation.40 Similar findings were observed with IL-13, which induces an increase in RhoA expression by aSMC.41 Those results suggest a potential role of Ca2+ sensitisation pathway dysfunction associated with asthma AHR.
Aside inflammatory cytokines, small molecules can also interact with the contractile apparatus such as nitric oxide (NO). In physiological conditions, NO is a potent bronchodilator through the production of cGMP by activated cytosolic guanylate cyclase leading to decreased intracellular Ca2+.42 Bronchial NO mainly derived from epithelial cells on the one hand and inhibitory non-adrenergic non-cholinergic nerve terminals.43 44 Inhibition of NO synthesis by L-NG-Nitro arginine methyl ester in vitro and in vivo in guinea pigs enhances the airway hyper-responsiveness in response to histamine.45 Though, its precise role in the context of asthma remains controversial.46 Indeed, inflammatory environment, especially type 2 driven, is a potent activator of inducible NO synthase of type 2 in epithelial cells.47 48 Exhaled NO measured in asthmatic patients is correlated with AHR.49 50 Association of NO with worsened AHR, despite its physiological bronchodilator effect, can be partly explained by collateral damages linked with peroxynitrite production and side effects on vessels with increased permeability and bronchial oedema.
Autonomous innervation of aSMCs is another modulator of AHR. TRPA1 channels are located at sensory nerves, predominantly on C fibres and is also expressed in non-neuronal cells including airway inflammatory cells, SMC, epithelial cells and fibroblasts.51 Activation of TRPA1 by environmental irritants such as cigarette smoke or air pollution leads to the activation of bronchopulmonary C fibres in an experimental model and is implicated in cough.52 Inhibition or knockout of TRPA1 channels leads to an inhibition of neuropeptide release and airway hyperreactivity in an ovalbulmin-challenged mice model of asthma.53 Whether such pathway is implicated in human asthma pathophysiology remains to be demonstrated.
aSMC beyond contraction: bronchial remodeling
Airway remodelling presentation may differ between groups of moderate-to-severe asthmatic patients in terms of aSMC mass and basement membrane thickening.54 Interestingly, airway remodelling can appear early in asthma, even in children.55 56 Significant bronchial remodelling in childhood, characterised by reticular basement thickening and increased aSMC mass, is associated with severe disease.57 Moreover, airway remodelling is also associated with persistent obstruction in severe asthmatic children.58 Those elements are in favour of an early role played by airway remodelling in asthma natural course.
Physiologically, aSMC presents low proliferation capacities and a contractile phenotype characterised by the expression of sm-α-actin, smooth muscle γ-actin, smooth muscle myosin heavy chain, calponin, h-caldesmon, SM22, smoothelin and metavinculin.59 Though, in inflammatory environment, aSMC have the ability to switch their phenotype with an increase of their proliferation and migration capacity leading to hyperplasia.60 Mitochondria biogenesis is also affected. In proliferative aSMC from asthmatic patients, increased mitochondrial mass and activity can be observed associated with altered calcium homeostasis.61
Phenotype switch can be induced in vitro by several growth factors and cytokines presented in table 1. Analysis of induced sputum in asthmatics showed that many of these molecules are indeed oversecreted.62 Bronchial inflammation associated with remodelling can be driven by the environment through the ability of bronchial epithelium to secrete alarmins, namely thymic stromal lymphopoietin (TSLP), IL-25 and IL-33, in response to harm.11 It has been demonstrated that human aSMC expressed receptor to TSLP and IL-25 and that their stimulation lead to proinflammatory and synthetic phenotypes.63 64 Conversely, it has been shown by air–liquid interface coculture that injured epithelial cells stimulate aSMC proliferation through the production of proinflammatory molecules (IL-6, IL-8, monocyte chemotactic protein-1) and matrix metalloproteinase-9.65 Mechanical stimulation of epithelial cells is another path leading to phenotype switch of aSMC. A significant proliferation of aSMC could be induced in vitro by compression of epithelial cells.66 Combination of inflammatory cytokines, such as type 2 cytokines, leads to complex modification of aSMC phenotype. For example, whereas IL-13 alone have an antiproliferative effect on cultured aSMC, it also increases the expression of CysLT1 receptor enhancing leukotriene induced proliferation.67 68 Interestingly, stimulated aSMC are also able to synthesise proinflammatory factors such as the platelet-derived growth factor (PDGF), fibroblats growth factor, IL-1β, transforming growth factor-β, IL-5, IL-6, IL-8, IL-17, which further amplifies phenotype switch.3
Mitogenic factors of aSMC
Composition of the extracellular matrix (ECM) itself also influences the phenotype and functions of aSMC.69 Laminin reduces the aSMC proliferation and increases the expression of contractile proteins such as sm-α-actin and smooth muscle myosin heavy chain.70 On the contrary, fibronectin promotes the aSMC proliferation but decreases the expression of contractile proteins.70 During asthma, the synthesis of laminin is reduced while the fibronectin synthesis is increased promoting the switch of aSMC to a proliferative phenotype.71 In response, aSMC participate to the deposition of the ECM through increased MMP-9 and MMP-12 expressions as it has been shown in bronchial biopsies from severe asthmatic patients.72
At a cell signalling level, aSMC proliferation is principally under the control of extracellular signal-regulated kinase (ERK) and PI3K pathways by increased expression of cyclin D1. ERK protein, a member of the MAPK familly, is a central regulator of cell cycle entry and G1 progression essential to aSMC proliferation.73 In parallel, Akt1, an effector of PI3K, inhibits the constitutively active glycogen synthase kinase 3 and an activator mTOR and p70 S6 kinase which are important for transcriptional activation and protein translation leading to aSMC proliferation and hypertrophy.74 PI3K can also activate Rac1 and Cdc42 in order to promote the cell proliferation thanks to cyclin D1.75 In addition, Rac1 forms part of the Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxydase complex and also participates in the reactive oxygen species production which is involved in the aSMC mitogenesis.76 We recently demonstrated that Rac1 was essential to the increased proliferation capacities of aSMC from asthmatic patients in comparison to controls in basal condition and after mitogenic stimulation.77 We identified the signal transducer and activator of transcription 3 as the main effectors involved in such Rac1-dependent mecanism. Interestingly, inhibition of Rac1 activity in a mouse model of asthma prevented aSMC hyperplasia. aSMC hyperplasia also results from the migration of aSMC or progenitors in response to mitogen factors such as the PDGF, the vascular endothelial growth factor, the transforming growth factor β (TGF-β) or IL-1β. The P38 MAPK, PI3K and ERK pathways are involved in migration as their inhibition leads to decreased aSMC migration due to reduced phosphorylation of heat shock protein 27 implicated in the F-actin polymerization necessary for cell motility.78 Moreover, the migration of aSMC in response to PDGF is significantly impacted by the inhibition of PI3K, ERK or ROCK pathway.79 80
Aside its proliferative capacity, activated aSMC also demonstrate improved capacity to interact with immune cells through increased expression of their surface molecules such as VCAM-1, ICAM-1, CD44 and LFA-1.81 Noteworthy, aSMC display closed interaction with mast cells and are able to synthetize powerfull mastocyte chemotactic agents such as the stem cell factor but also TGF-β1 and tumour suppressor in lung cancer‐1.82 In parallel, aSMC express both CD44 and CD51 which are involved in the mast cell adhesion.83 Interaction of mast cells with aSMC promotes the degranulation and cytokines production by mast cells ultimately leading to aSMC contraction and proliferation.84 aSMC can also interact with other immune cell types such as T cells via CD44, which induces DNA synthesis of aSMC and promotes its proliferation.85
As presented, aSMC has important impacts on asthma pathophysiology not solely due to their contractile activity but also to their ability to interact with other cell types leading to complex remodelling activity. Such a broad involvement makes the clinical evaluation of its action at a patent level complex, as it is discussed in the next section.
Bronchial smooth muscle in asthma clinical practice
In routine practice, evaluation of aSMC’s function cannot be fulfilled directly. Indeed, conventional pulmonary function tests, such as spirometry and plethysmography only give access to an overall sight of their implication in airway obstruction. New tools currently developed to further assess new facets of its function are described below.
Airway obstruction in asthma
Airway obstruction is necessary to asthma diagnosis along with relevant and consistent respiratory symptoms. Pulmonary function tests in asthma, particularly forced spirometry, are standardised and aim to prove such obstruction.86 Evolution of obstruction through time is critical considering asthma care. Data from birth cohorts showed that children with low forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) ratio had a steeper slope of evolution of FEV1/FVC ratio through time until adulthood that increases the risk of developing asthma.87 In parallel, patients self-reporting asthma experienced a faster decline of FEV1 through time than healthy volunteers in a 15-year prospective study performed in Denmark.88
Bronchodilators response tests are available to assess the role of bronchial smooth muscle contraction in obstruction for an individual patient.86 However, negative bronchodilator test does not imply that aSMC are not relevant in asthma symptoms. Indeed, a fixed obstruction (FEV1/FVC ratio under lower limit of normal values and/or FEV1 under 80% of predicted value after bronchodilators) can appear in about 20% of never ever-smoking adult asthmatic patients after 10 years of follow-up.89 Such persistent airway obstruction had been linked with increased airway smooth muscle area in a population of severe asthmatics under standardised high dose anti-inflammatory treatment.90 Though, airway remodelling is not only dependent on aSMC. It has been shown recently by cluster analysis of pathological examination of bronchial biopsies from asthmatic patients and healthy individuals that bronchial remodelling could be classified into several groups depending on the component involved (bronchial smooth muscle, basal membrane).54
Considering small airways impairment in asthma, available explorations are currently imperfect. A study highlighted that mid-expiratory and instantaneous flows (FEF25-75 and FEF75) did not significantly add informations to FEV1 and FEV1 to FVC ratio.91 Use of impulse oscillometry and nitrogen breath washout technics identified around 1/3 of asthmatic patients displaying markers of small airways dysfunction.92 Though, data clearly linking pathology with oscillometric data are still lacking. Conversely, specific imagery approaches are currently assessed. Non-invasive evaluation of obstruction by hyperpolarized 3HE MRI showed that ventilation defects due to obstruction often persisted in time and location under stable or provoked (methacholine) conditions in a small series of patients.93 Interestingly, markers of ventilation heterogeneity linked to small airways involvement inversely correlated with variation of asthma control score under inhaled corticosteroid treatment of asthmatic patients.94 Nevertheless, MRI lacks availability needed for clinical practice.
Airway hyper-responsiveness in asthma
AHR is an important argument in favour of asthma diagnosis that can be sought by direct and indirect provocation bronchial tests.95 96 Methacholine and histamine tests are the principal direct provocation tests available. They aim at triggering direct bronchial smooth muscle contraction through inhalation of determined cumulative doses of stimulant. However, this test explores only selected pathways of bronchial smooth muscle contraction to the exclusion of the others described in the previous sections. In addition, its overall sensitivity is around 60%–90% and specificity around 90%.97 Its use in clinical practice is then principaly reserved to intermediate probability of asthma at diagnosis and is not recommended for follow-up.
Indirect tests mainly include exercise-induced and eucapnic hyperventilation tests. The objective of such tests is to provoque a deshydratation of respiratory airways that stimulates the secretion of various cytokines and inflammatory mediators by bronchial epithelium and sub-mucosa triggering hypersensitive aSMC contraction.96 Exercise-induced bronchoconstriction is associated with asthma with a good specificity but low sensitivity. In an overall population, exercise-induced bronchoconstriction under standardised exercise correlated with airflow limitation (FEV1) but also with age and sex.98 Noteworthy, some patients displaying significant positive indirect AHR tests would not react to direct stimulation of airway smooth muscle contraction.99
Unmet needs in aSMC functional evaluation in asthma
Diagnostic tools determining the exact role played by bronchial smooth muscle in asthma at an individual level need to be developed. Indeed, spirometry isn’t sufficient to precisely incriminate the responsible agent (inflammation, bronchial smooth muscle, infection triggers, combination of those). Tests specifically exploring bronchial contractility and its determinants could potentially be of interest in order to guide therapy, particularly concerning the use of long-acting bronchodilators. Conversely, it would be interesting to evaluate the proliferative activity of bronchial smooth muscle in order to early detect and prevent bronchial remodelling. Finally, the improvement of physiopathological knowledge could lead to the development of new targeted therapeutic strategies alongside their specific biomarkers.
aSMC as a target in asthma
Conventional therapies: inhaled and oral treatments
Conventional therapies in asthma principally target 2 pathophysiological mechanisms: inflammation and bronchoconstriction. Inhaled bronchodilators (beta-2 agonist and anticholinergic) directly target aSMC by decreasing its contractility in order to improve airflow and limit chronic and acute symptoms. Though, pharmacological researches mainly focused on the improvement of the length and/or delay of action. Interestingly, a proof-of-concept clinical trial showed in a small cohort of severe asthmatics (31 patients) that gallopamil, a calcium ion channel inhibitor, could reduce the aSMC bronchial area and thickness after 1 year of treatment in comparison with baseline associated with reduced exacerbation after the end of treatment.100 However, further clinical trials were withdrawn by the pharmaceutical companies.
Even if inhaled corticosteroids target principally inflammatory effectors, it also affects aSMC contractility and proliferation. Glucocorticoids reduce the expression of α-smooth actin and the short isoform of MLCK by aSMC in response to TGF-β which dampens its contractility.101 It also decreases the expression and phosphorylation of CPI-17 by aSMC in a rat model leading to lower MLC phosphorylation and improved AHR.102 Furthermore, ciclesonide effectively reduced key bronchial remodelling features, such as goblet cell hyperplasia or immune reactive aSMC, in a rat model of asthma.103 Considering bronchial remodelling, high doses of inhaled steroids also improved the submucosal hypervascularity but also the basement membrane thickness in small clinical studies.104 105
Biotherapies and aSMC
No biotherapy directly targeting aSMC is currently under clinical development to our knowledge. However, available biotherapies that target inflammation can have implication in aSMC (table 2).
Effect of approved biotherapies on pulmonary function tests in allergic and eosinophilic severe asthma (phase III trials)
Omalizumab is a humanised monoclonal antibody targeting immunoglobulin (Ig) E to treat severe allergic asthmatic patients. Phase III study demonstrated that omalizumab significantly improves asthma exacerbation rate compared with placebo.15 106 It also showed that omalizumab could slightly improve morning peak expiratory flow and FEV1. Monoclonal antibodies targeting IL-5 pathway namely mepolizumab, reslizumab and benralizumab are now approved to be used to treat severe eosinophilic asthma.107 Considering improvement of prebronchodilator and postbronchodilator FEV1, biotherapies targeting IL-5 reached statistic significance overall, though clinical significance remains questionable (about+100 mL vs placebo). Conversely, dupilumab, a fully human monoclonal antibody targeting IL-4 receptor α also demonstrated a significant reduction of exacerbation rate and efficient oral corticosteroid tapering in patients suffering of uncontrolled moderate to severe asthma.17 108 Interestingly, a significant improvement of FEV1 under treatment with dupilumab was observed at 24 weeks of treatment with mean difference above 200 mL.108 In a real-life asthma cohort, dupilumab also improved FEV1 by 10% (predicted values) after 1 year of treatment.109 Recently, a treatment with tezepelumab, a human monoclonal antibody targeting the TSLP, lead to a 130 mL increase of pre bronchodilator FEV1 in comparison with the control group.110 Such improvements, to be confirmed in the long term, tends to underline the importance of the interactions between inflammation and bronchial smooth muscle and the interest in simultaneously targeting multiple actors.
Few trials specifically studied the effect of biotherapies on pulmonary functions. Benralizumab failed to significantly improve prebronchodilator FEV1 and hyperinflation in SOLANA trial.111 To note, this trial aimed at assessing benralizumab efficiency at short-term (84 days) and potentially lack interesting effects on obstruction through prolonged treatment. Small-sized clinical studies have shown that mepolizumab could possibly improve small airway function evaluated by multiple-breath nitrogen washout test and forced oscillometry after a few months of treatments.112
Bronchial thermoplasty
In order to specifically target bronchial wall including aSMC, an interventional endoscopic technic was developed: bronchial thermoplasty. Its objective is to lower the airway wall thickness by direct thermic energy application. In AIR-2 clinical trial, bronchial thermoplasty significantly improved quality of life with a trend in favour of better asthma control through lower exacerbation rate in comparison with the sham group.113 Good long-term tolerance has been shown after 5 years of follow-up.114 Considering bronchial remodelling, in a small prospective case series (n=11), bronchial thermoplasty slightly decreased the airway wall thickness and air-trapping 2 years after the procedure, even if no significant improvement of airway lumen could be observed.115 However, no significant effect had been observed on small airways evaluated by oscillometry 6 months after the procedure, despite significantly improved clinical markers.116 Interestingly, a proof-of-concept pilot study found that bronchial thermoplasty could nevertheless improve dynamic hyperinflation in selected patients.117 Though, such results need to be confirmed in larger and control clinical studies.
Data about bronchial thermoplasty effects on airways physiology and asthma pathophysiology are now available. aSMC area (α-SMA staining) significantly decreased in short term (6 weeks) after bronchial thermoplasty consistent with lower smooth muscle mass.118 The TASMA randomised clinical study confirmed such data by including a parallel delayed bronchial thermoplasty group of severe asthmatic patients as controls.119 Nevertheless, modification of aSMC mass did not correlate with improvement of asthma control and related quality of life scores (Asthma Control Questionnaire, Asthma Quality of Life Questionnaire). In parallel, it was recently reported that bronchial thermoplasty induced a decrease of mucus production assessed by MUC5AC epithelial expression at 12 months post procedure.120 Bronchial thermoplasty can also modify cell cross-talk between the different components of airway wall. It has been reported that it blocked the production and secretion of heat-shock protein-60 by the epithelium that triggered in part remodelling in asthma by fibroblasts.121
Unmet needs in targeted treatment of aSMC in asthma
Few innovative strategies targeting specifically aSMC are available. Although bronchial thermoplasty has shown interesting results, this technique remains complex and reserved to a limited number of patients. Besides, in an era of precision medicine, tools that predict response to a treatment strategy in an individual setting are essential. Few studies are available concerning such biomarkers to predict efficacy of bronchial thermoplasty. Fixed or reversible obstruction status was not significantly associated with clinical response to bronchial thermoplasty.122 Small molecules that directly target key signalling pathways implicated in aSMC contraction and proliferation could be an interesting therapeutic opportunity. For example, inhibition of Rho and Rac activation could potentially reduce airway hyper-responsiveness and remodelling. Nevertheless, those have long been considered as undruggable due to their ubiquitous expression and critical biological roles that could lead to serious side effects. The identification of tissue and condition specific regulators of Rho superfamily activation, such as guanine exchange factors, could be a promising way to overcome such limit.
Conclusion
Asthma represents a broad-spectrum disease involving at different levels epithelial dysfunction, sustained bronchial inflammation and bronchial smooth muscle dysfunction. Knowledge about bronchial smooth muscle role in asthma pathophysiology had been considerably improved (figure 3). Mechanisms of bronchial contraction are better understood and new intracellular pathways had been discovered. However, aSMC should not be only considered as simple contraction actors. Indeed, implication of aSMC airway remodelling and their secretion capacities are far more important than previously expected.

{kind=link}
{kind=link}
{kind=link}
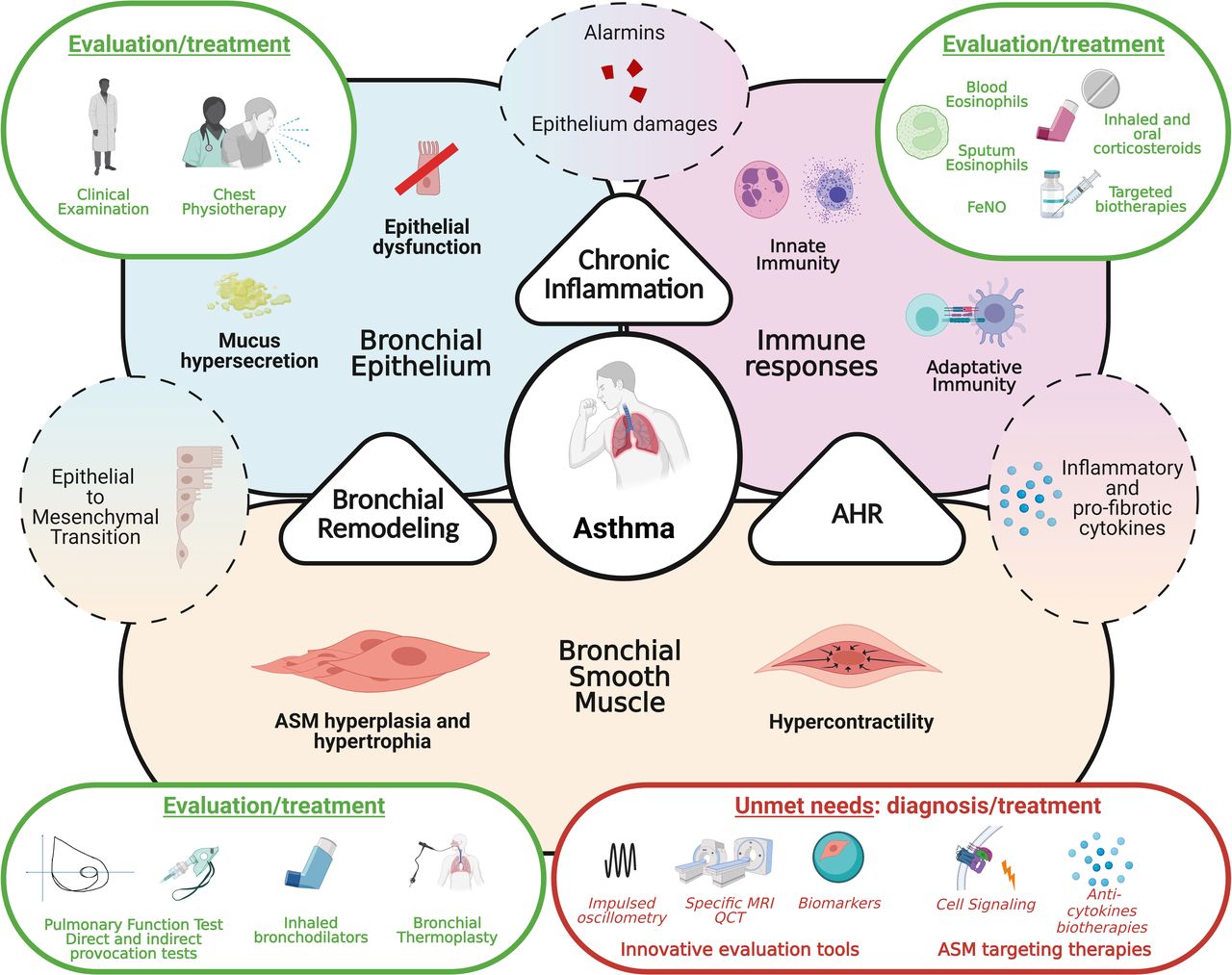
Bronchial smooth muscle cells in asthma from pathophysiological consideration to evaluation and treatments. Asthma development relies on three main pathophysiological processes, bronchial remodelling, chronic inflammation and airway hyper-responsiveness, consequences of alteration of bronchial epithelium and ASM and development of inadequate immune responses. Whereas anti-inflammatory therapeutic strategies got significantly improved in the last years, little progress has been made considering ASM. Innovative precise evaluation tools of ASM function along with specific targeting strategies need to be developed. AHR, airway hyper-responsiveness; ASM, airway smooth muscle; FeNO, exhaled fraction of azote monoxide.
In routine practice, aSMC functions are mainly assessed through pulmonary function tests and provocation tests. Innovative tools such as forced oscillometry and dedicated imaging are used in clinical research but did not reach clinical practice yet. In parallel, improvement of basic sciences comprehension of airway smooth muscle biology doesn’t lead to real targeted strategies for now. On the one hand, bronchodilators developed for the last decades mainly targeted the same pathways (adrenergic and muscarinic receptors) and on the other hand, bronchial thermoplasty still lacks predictors of success even though promising results have been described. Development of tools to better assess aSMC activity in clinical practice along with new targeted treatment are mandatory to identify patients for whom SMC dysfunction is preponderant and need to be specifically treated.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
DH and LR contributed equally.
Contributors DH, LR and VS drafted the manuscript. DH and LR drew the figures. F-XB, AM, GL and VS critically revised the manuscript. All authors gave their final approval for publication.
Funding This work was supported by a grant from the French Regional Council of Pays de la Loire, IRSR-PL project StaRac. DH is supported by a scholarship from Foundation pour la Recherche Médicale, poste pour internes et assistants program, FDM201906008829.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.