
Bacterial Adaptation during Chronic Respiratory Infections

Centre of Microbial Host Interactions, Institute of Technology Tallaght, Dublin 24, Ireland
*
Author to whom correspondence should be addressed.
Pathogens 2015, 4(1), 66-89; https://doi.org/10.3390/pathogens4010066
Submission received: 3 December 2014
/
Revised: 15 February 2015
/
Accepted: 25 February 2015
/
Published: 2 March 2015
(This article belongs to the Special Issue Respiratory Pathogens)
Abstract
:Chronic lung infections are associated with increased morbidity and mortality for individuals with underlying respiratory conditions such as cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD). The process of chronic colonisation allows pathogens to adapt over time to cope with changing selection pressures, co-infecting species and antimicrobial therapies. These adaptations can occur due to environmental pressures in the lung such as inflammatory responses, hypoxia, nutrient deficiency, osmolarity, low pH and antibiotic therapies. Phenotypic adaptations in bacterial pathogens from acute to chronic infection include, but are not limited to, antibiotic resistance, exopolysaccharide production (mucoidy), loss in motility, formation of small colony variants, increased mutation rate, quorum sensing and altered production of virulence factors associated with chronic infection. The evolution of Pseudomonas aeruginosa during chronic lung infection has been widely studied. More recently, the adaptations that other chronically colonising respiratory pathogens, including Staphylococcus aureus, Burkholderia cepacia complex and Haemophilus influenzae undergo during chronic infection have also been investigated. This review aims to examine the adaptations utilised by different bacterial pathogens to aid in their evolution from acute to chronic pathogens of the immunocompromised lung including CF and COPD.
1. Introduction
The respiratory tract is the first point of contact in the lungs for pollutants and microorganisms, with, on average, 10,000 L of air inhaled per person per day. In healthy individuals, infections are rare despite bacteria being inhaled frequently, due to sophisticated host defence mechanisms at the lung mucosa. The combination of cilia and the mucous gel film on the epithelial surface allows for entrapment and clearance of particles and bacteria in healthy lungs. Antimicrobial peptides produced by epithelial cells and local immune responses such as secretory IgA and resident airway macrophages all contribute to the prevention of bacterial colonisation [1]. The healthy respiratory system has its own micro-flora, including Staphylococcus epidermidis and Cornyebacteria, S. aureus, non-haemolytic and alpha-haemolytic Streptococci and Neisseria species with occasional carriage of Streptococcus pneumoniae and H. influenzae in the upper airways [2]. The lower airway micro-flora, previously considered sterile, is now known to be colonised with a diverse range of genera including Pseudomonas, Prevotella, Streptococcus, Fusobacteria, Veillonella, Haemophilus and Acinetobacter species [3]. There are varying reports of the microbial communities in healthy lungs, and research into the respiratory microbiome in both healthy and disordered states is ongoing [4]. Understandably, the resident microbiome will depend on geography, climate and other environmental conditions [5].
Both cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) are characterised by airway inflammation, altered mucus production and diminished mucocillary clearance. COPD and CF can cause bronchiectasis and both of these disorders are characterised by repeated cycles of inflammation, tissue damage and chronic bacterial infections contributing to a rapid decline in pulmonary function [6]. In a comparison of the COPD and healthy lung microbiome, the COPD state was found to differ in the levels of certain genera including Afipia, Brevundimonas, Curvibacter, Moraxella, Neisseria, Corynebacterium, Undibacterium, Capnocytophaga and Leptolyngbia [3]. This is in agreement with a comparison of the microbiomes in COPD and asthmatic patients with healthy controls, highlighting a diverse population of bacteria in the healthy controls. The composition of the microbiome of the healthy controls was considerably different from the diseased states [7]. The absence of functional cystic fibrosis transmembrane regulator (CFTR), a chloride ion channel, in CF patients results in airway surface liquid depletion [8]. CF sputum has a lower viscosity in comparison to asthma or bronchitis sputa but is highly tenacious leading to a reduction in cough clearance of infected phlegm, and subsequently inducing an inflammatory state in the airways [9]. A significant reduction in diversity of bacteria is also a hallmark of the CF microbiome. Older patients exhibit poorer lung function with more uneven and less diverse, more specialised communities, compared to younger healthy patients [10].
Alterations in the CF lung surface provide an environment which is exploited by CF pathogens. P. aeruginosa is the most prevalent pathogen that colonises people with CF. Another pathogen that chronically colonises the CF lung of predominantly adult patients is Burkholderia cepacia complex (Bcc), a group of 18 phenotypically similar, genetically distinct Gram negative bacterial species. The two most clinically relevant species are B. cenocepacia and B. multivorans. Other pathogens associated with CF lung infection include, Haemophilus influenzae, Stenotrophomonas maltophilia, Achromobacter xyloxidans, genus Pandoraea and Gram positive species including S. aureus, and Streptococci as recently reviewed [11]. These lung pathogens chronically colonise the CF lung contributing to gradual but unrelenting decline in pulmonary function and tissue damage [12]. COPD is characterised by restricted expiratory airflow from numerous anatomical lesions, including loss of lung elasticity, fibrosis and narrowing of the small airways. Inflammation, oedema, and secretions also contribute variably to airflow limitation. Smoking is a significant factor in COPD, inducing inflammatory responses, including oxidants and proteases, which play a major role in lung damage. In addition, cigarette smoke modifies lung repair responses in several ways [6]. Non-typeable H. influenzae is the most common coloniser of the COPD lung with Moraxella catarrhalis and P. aeruginosa being identified to lesser extents. H. influenzae is associated with persistent chronic infections, in contrast to M. catarrhalis, which is generally cleared within one month of acquisition [13]. P. aeruginosa, which chronically colonises the CF lung, is not frequently associated with persistence in COPD patients [14].
2. Bacterial Adaptation
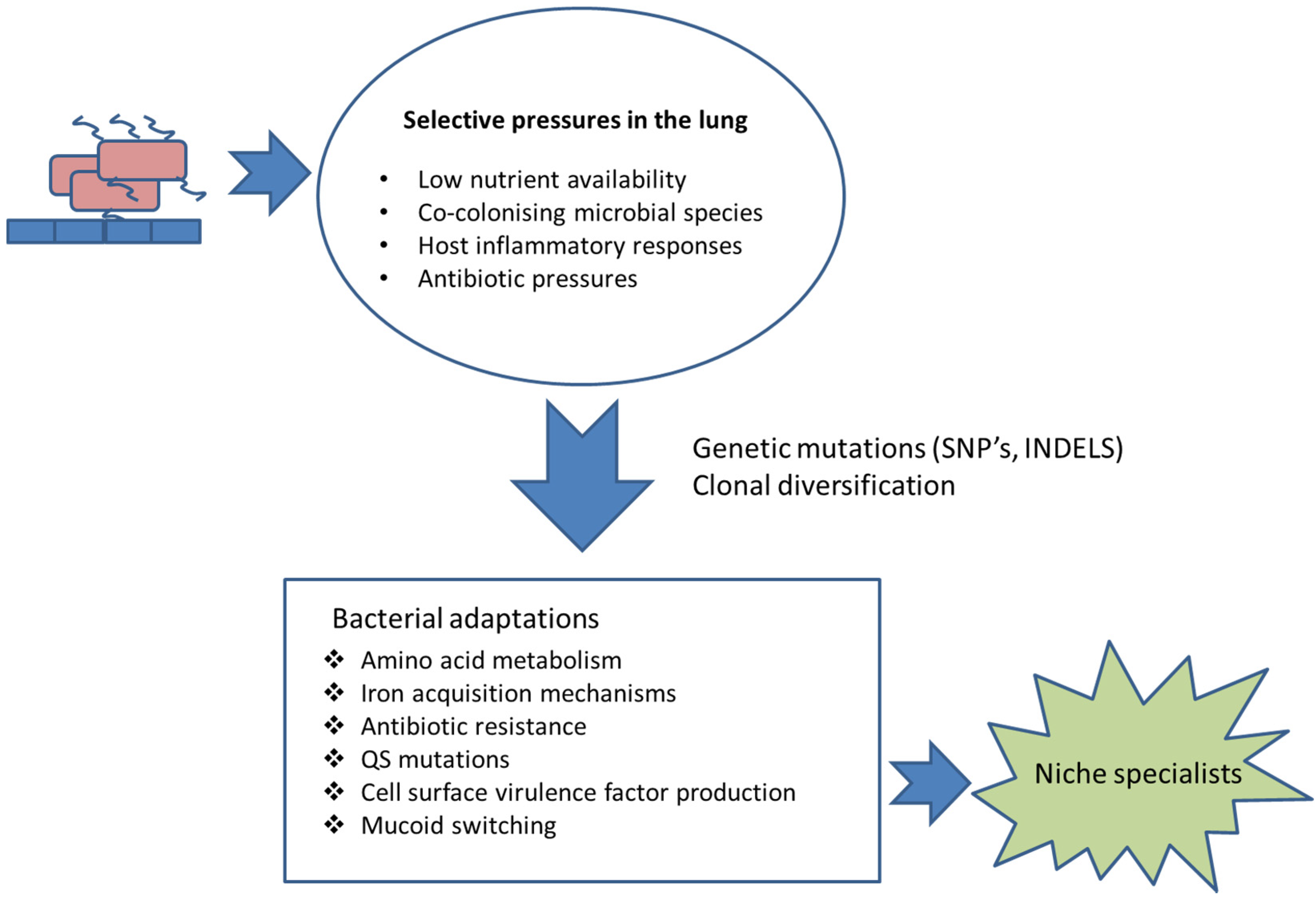
Virulence factors or determinants are often non-essential to the pathogen but, if lost, they inevitably result in attenuated virulence [15]. Bacterial virulence factors are commonly in direct contact with the host or are involved in concealing the pathogen from the host immune system [16]. For example, outer membrane proteins (OMPs) are essential in adhesion, colonisation, intra-cellular invasion, antimicrobial resistance and intracellular communication processes [17]. The proteins can be considerably immunogenic [18]. In addition, the polysaccharide cell wall and capsule have anti-phagocytic properties required for immune evasion. Microbial secretory proteins are involved both in host microbe interactions and in tissue damage. The cell wall and outer membrane moieties such as lipopolysaccharide (LPS) are involved in recognition and host inflammation [16]. It is inevitable that in order for microbial pathogens and populations to chronically colonise a host, they need to adapt to the host environment and evolve over time within the host (Figure 1). The two key drivers for this are the enhanced survival and proliferation of the adapted pathogen to the host environment together with avoiding detection and destruction by the host immune system. Many, but not all, pathogens persist in the lung due to a down regulation of virulence factors and pathogen associated molecular patterns (PAMPS) to avoid detection by the host (Table 1). This review will focus on adaption of pathogens to the host lung during chronic colonisation. The majority of the research has focused on CF-associated pathogens; however, adaptations of bacteria associated with COPD have also been included.

{kind=link}
Table 1.
Bacterial adaptation during chronic infection: examples of adaptations and the general outcome during chronic respiratory infections.
| Bacterial Component | Pathogen | Adaptation | Effect of Adaptation | Disease | References |
|---|---|---|---|---|---|
| Genome | P. aeruginosa & S. aureus | Mutations (single nucleotide polymorphisms, insertions, deletions) | Non-synonymous mutation effecting gene product | CF | [19,20] |
| Outer membrane proteins | B. cenocepacia | Upregulation of siderophore interacting protein, pyochelin receptor FptA, and TonB receptor | Iron chelating | CF | [21] |
| P. aeruginosa | Phu mutations | Increased transcription and switch towards haemoglobin utilisation for iron | CF | [22] | |
| H. influenzae | Alteration of outer leaflet | Immune evasion | COPD | [23] | |
| Lipopolysaccharide | P. aeruginosa | Loss of O-antigen | Immune evasion | CF | [24] |
| B. cenocepacia | Reduced expression of enzymes involved in O-antigen and lipid a synthesis | Modifications in LPS and LPS expression | CF | [25] | |
| Exopolysaccharide | P. aeruginosa | Non-mucoid to mucoid switch | Reduced virulence factor expression, poorer lung function in patients | CF | [26] |
| B. multivorans | Mucoid to non-mucoid switch | Reduced in vivo virulence, reduction in virulence factor expression | CF | [27] | |
| B. cenocepacia | Mucoid to non-mucoid switch | Poorer outcome in patients | CF | [28] | |
| Motility | P. aeruginosa | Loss in flagella associated motility | Phagocytosis evasion | CF | [29] |
| P. aeruginosa | Loss of swimming, swarming and twitching | Loss of motility | COPD | [30] | |
| B. cenocepacia | Upregulation of flhB, flhF, fliG, fliK, fliJ, fliN, flgB, flgD, flgE, flgF, flgG, flgH, flgI | Increase flagellin assembly | CF | [31] | |
| Colony morphotypes | S. aureus | Small colony variants | Antibiotic resistance, intracellular survival, reduction in α-toxin expression | CF | [32] |
| P. aeruginosa | Rugose small colony variants | Increased auto-aggregative properties, increased biofilm formation | CF | [33] | |
| B. cenocepacia | Shiny colony variant | Reduced virulence and biofilm formation | CF | [34] | |
| Quorum sensing | P. aeruginosa | lasR and rhlR mutation | Reduced production of QS associated virulence factors, increased resistance to β lactams, growth advantage with low levels of amino acids | CF | [35] |
| S. aureus | agr mutation | Attenuated virulence, non-haemolytic | CF | [36] |
Figure 1.
Selection and adaptation. Examples of selective pressures to which chronically colonising respiratory pathogens are exposed and the adaptations that they undergo, in order to enhance chances of survival.
Figure 1.
Selection and adaptation. Examples of selective pressures to which chronically colonising respiratory pathogens are exposed and the adaptations that they undergo, in order to enhance chances of survival.

2.1. Bacterial Genomic Evolution in the Host
The microbial genome evolves by a combination of point mutations, gene conversions and rearrangements, insertion of foreign DNA and deletions, enabling bacteria to adapt to the host environment. In addition, within an individual host, a clonal isolate can diverge forming separate, evolutionarily diverse species. Furthermore, the genetic information can be shared between pathogens via a horizontal gene transfer mechanism, further facilitating the adaptation of these opportunistic bacteria to their environments [37].
Investigations of the genomes of sequential isolates indicate that both P. aeruginosa and S. aureus are associated with reduced virulence over time of colonisation. In a shotgun whole genome sequencing study of P. aeruginosa sequential isolates from a CF patient over the course of eight years, it was clear that the isolates evolved within the host by a process of natural selection to reduce expression of virulence factors. There was a higher ratio of non-synonymous to synonymous mutations associated with an alteration in biological function. Mutations in genes regulating O-antigen biosynthesis, type III secretion, twitching motility, exotoxin A, multidrug efflux, osmotic balance, phenazine biosynthesis, quorum sensing (QS) and iron acquisition were all evident in the eight year isolate relative to the early isolate. The later isolate also expressed a mutation in mutS, a DNA mismatch repair gene, associated with increased mutation rates, which is potentially beneficial to the pathogen during long term colonisation [19].
In an attempt to determine the genetic within-host diversification S. aureus undergoes in the CF lung, a set of three sequential isolates, which were deemed identical based on sequence type, were examined over a period of 23 months. During the course of infection, 23 point mutations and 15 indels were identified; with 95.7% being non-synonymous mutations, consistent with niche adaptation of pathogens in CF. Mutations in fusA, associated with fusidic acid resistance was noted in the later isolate. In addition to this, the gene encoding the branched chain amino acid binding pocket of the global regulator CodY, normally involved in repressing cell virulence, contained a non-synonymous point mutation in this later isolate [20]. This later isolate had an increased ability to grow in threonine deficient media in comparison to the early and middle isolates, which has previously been associated with CodY mutants and is likely to be beneficial in vivo [38]. Multiple independent indel polymorphisms in all three isolates affecting SigB activity, a regulator of stress response, were also observed [20]. The parallel adaptation of mutations that influence virulence factor expression indicates a strong selective pressure on these loci contributing to a beneficial phenotype in vivo. In addition, a variation in the accessory genome in the later isolates was observed. Two distinct phage insertions were present in the initial isolate which encode secreted virulence factors, enterotoxin A and staphylokinase. The middle isolate and late isolates each lost one of these factors, illustrating a dynamic relationship between both the core and accessory genomes of S. aureus in suppressing virulence during chronic infection [20].
2.2. Diversification
While unique strains can be acquired during the early stages of infection, chronic colonisation is generally due to persistence of bacterial strains of the same lineage. The process of adaptation most likely occurs, one cell at a time, giving rise to sub-populations within the host. If these subpopulations can co-exist, then diversification of the population is inevitable. It is widely accepted that there is considerable heterogeneity and diversity across the P. aeruginosa population within the lungs of an individual with CF. This can be driven by a range of pressures including antibiotic treatment. Fothergill et al. [39] have shown significant fluctuations in morphotypes, virulence factors and antibiotic susceptibility within the P. aeruginosa populations during exacerbations and antibiotic treatment. In addition, genotypic variability arose within the P. aeruginosa population of each individual’s lung, predominantly due to phage activity. Variability in genotypes had also been reported by Smith et al. [19] in their earlier study of CF P. aeruginosa isolates over eight years, with as many as five genotypes (all from the same lineage), being identified in one sample from a three-year old patient. The extent of this diversification within a patient is highlighted by recent work by Workentine et al. [40] in their study of 169 isolates from a single patient. All isolates belonged to a single clonal group by pulse-field gel electrophoresis (with only two or three band differences), yet a high degree of phenotypic variability was observed even within isolates of the same colony morphotypes. A study of COPD colonised patients demonstrated that diversification of P. aeruginosa is not unique to CF patients and that different coexisted with different antibiotic susceptibilities within the individual patients for the entire period of the study [30]. Another study of COPD patients over 10 years showed that a minority of patients were chronically colonised with the same P. aeruginosa clone, but among these clonal diversification was evident [41].
Diversification is not exclusive to P. aeruginosa infection and has also been observed in Bcc colonised patients. A series of sequential clonal isolates recovered from a B. multivorans colonised patient colonised for 13 years showed diverse morphologies across the population [27]. B. dolosa, a relatively rare but considerably virulent member of Bcc showed distinct morphotypes within the same sputum sample [42]. Clonal isolates of B. cenocepacia isolated over 3.5 years of colonisation have also been shown to have diverged over time [21,25].
It has been recently demonstrated that adaptive mutations rarely fix within a patient’s population and that co-existence of diverse patients can exist for many years, adapting under the pressure of natural selection [43]. In an in-depth study of five CF patients chronically colonised with B. dolosa, Lieberman et al. showed that diverging populations coexisted for at least five years and that the diversification was driven by multiple adaptive mutations in the same genes evolving in parallel within an individual [43]. Diversification offers an insurance that facilitates persistence of the community as a whole during changes in environmental conditions [44].
2.3. Hypermutability
Hypermutable microbes have an increased spontaneous mutation rate, as a result of defects in the DNA repair system or proof reading systems. These mutators play a major role in the evolution of the pathogen over time [45]. Antibiotics select for these variants, as these undergo more genetic mutations and are better able to adapt and survive under the antimicrobial pressures in vivo [46]. It has also been shown that the host environment can also select for mutator microbes, within P. aeruginosa, S. aureus and H. influenza populations [45]. The hypermutable strains of H. influenzae are more commonly isolated from the CF host than non-CF patients, which is most likely due to the high levels of antimicrobial therapy administered to these individuals. Nevertheless these mutator strains confer a long term persistence advantage [12,47]. Interestingly these mutators are rarely isolated in acute infections [45].
Hypermutable P. aeruginosa strains become more frequent in later stages or chronic infection. Mutations in mutS, mutL and uvrD genes encoding proofreading proteins which normally correct errors during DNA replication give rise to these hypermutable strains [12]. It is important to note that the increase in hypermutable P. aeruginosa isolates later in CF patients suggest that genetic and phenotypic diversification plays an essential role in the adaptation of P. aeruginosa to the hostile and diverse CF lung environment and plays a role in survival in vivo by selecting for less virulent phenotypes [48].
Yang et al. carried out a comprehensive whole genome sequencing analysis of the transmissible P. aeruginosa DK2 lineage over a period of 35 years (200,000 bacterial generations) in CF patients in vivo [49]. From this extensive analysis of sequential isolates an initial period of niche adaptive evolution was evident. Over a 35 year period a total of only 180 SNP’s occurred. The later isolates may be dominated by negative selection which has lasted and remained within the lineage. The authors further state that the resulting success of this lineage and its ability to thrive and survive long term in vivo, and its ability to spread from patient to patient may be explained by the adapted evolutionary mechanisms employed by the pathogen [49,50].
3. Adaptations in Surface Molecules
3.1. Adaptations in Outer Membrane Proteins
The outer membrane proteins (OMPs) are key players for within-host adaptive strategies of these Gram negative bacteria. Many OMP’s are regulated by environmental factors, some of which enhance the adaptability of bacterial pathogens to the host environment and during the infection process. The majority of OMPs are composed of monomeric or trimeric barrels comprised of 8–22 anti-parallel β-strands [17]. Membrane proteins that are involved in iron uptake are necessary for survival and virulence. Bacteria possess the ability to adapt to the low levels of iron in the host lungs by production of siderophores or iron chelating compounds which cleaves the hosts iron and utilises it as a co-factor. These are generally upregulated during chronic infection in vivo, in iron sparse environments. Proteomic expression studies suggest that up regulation of these iron chelating compounds by B. cenocepacia increases over time in the lung [21]. Deletions of these proteins in B. cenocepacia showed reduced virulence in vivo implicating the role of these proteins in virulence [51,52]. In terms of evolutionary advantage, enhanced survival via iron acquisition must overcome the potential risk of increased host detection. Active outer membrane transporters such as TonB and TonB dependent proteins also have a siderophore uptake function in the iron limited host environment, for example FpvA in P. aeruginosa [17,53]. Promoter mutations in the phu system which encodes the OM receptor, PhuR, the periplasmic soluble receptor, PhuT, and the permease, PhuUV, were identified in sequential clinical P. aeruginosa isolates [22]. This allowed increased transcription of these genes and an adaptation towards haemoglobin utilisation within the host.
The cell capsule OMPs have been implicated in serum resistance in the host, providing the bacterium with a means to evade the complement mediated killing. Nakamura et al. hypothesised that H. influenzae adapts to inflammation encountered during infection of the lower respiratory tract in COPD patients by modulation of its outer leaflet through increased expression of vacJ and yrb genes in order to minimize recognition by bactericidal anti-oligosaccharide antibodies [23].
3.2. LPS
LPS is an important glycolipid PAMP embedded in the outer cell leaflet of Gram negative bacteria. It induces a variety of responses in the host upon binding to TLR4, TLR2 and CD14 including pyrogenicity, platelet aggregation and induction of cytokines [54,55,56]. LPS composition has also been implicated in serum resistance [57]. LPS has three components: the toxic, highly acylated Lipid A; the central core oligosaccharides containing unusual sugars and the O-antigen. LPS composition increases asymmetry in membrane architecture, and the subsequent cross linking of LPS with divalent cations decreases the permeability to hydrophilic agents which increase the level of innate antibiotic resistance to these agents [58].
During chronic CF infection P. aeruginosa LPS structure is altered, with the loss or diminished production of O-antigen being frequently reported in later chronic stages of the disease [24]. These adaptations have recently been reviewed by Hauser et al. (2011) [12]. The immuno-stimulatory properties associated with the O antigen confer a selective advantage to cells lacking this structure. The fact that P. aeruginosa blood isolates are not frequently reported (in contrast to Bcc) could be as a result of the O-antigen structure alteration during chronic infection [59]. Proteomic profiling using 2-D DIGE studies have shown that O-antigen biosynthesis is also reduced in later sequential B. cenocepacia isolates. Both the expression of ManB (a phosphomannomutase) and an NAD-dependent epimerase, involved in lipid A and O-antigen synthesis, respectively, were lower in the later isolate taken close to patient death relative to an early isolate [21]. Two forms of the outer membrane assembly factor YaeT which is involved in maintaining a homeostatic LPS ratio was also up-regulated in the later isolate. This suggests that like P. aeruginosa, B. cenocepacia isolates can reduce the production of the immuno-stimulatory O-antigen and lipid A composition of the LPS in the later stages of infection, which could potentially benefit the pathogen in vivo [21].
3.3. Exopolysaccharide (EPS)
A mucoid phenotype is one of the adaptations that some pathogens undergo during respiratory infection. Mucoid colony morphology in P. aeruginosa most commonly occurs by overproduction of EPS alginate, a polymer of d-manuronic acid and l-glucuronic acid [60]. During initial infection of the CF lung, P. aeruginosa isolates are typically non-mucoid while later isolates are more mucoid in morphology as a result of mutations in the mucA gene encoding an inner membrane associated anti-σ-factor [26]. MucA normally limits the expression of the algD operon, encoding the enzymes required for alginate production and numerous stress responsive virulence genes by sequestering the alternative anti-σ22-factor encoded by algU [61]. Mutated mucA leaves this anti-σ22 free to activate many genes including genes involved in alginate production. This conversion in gene expression is controlled by the algU/algT regulon [62]. Remarkably the activation of this regulon also causes synchronised down regulation of central metabolism, motility and virulence and a simultaneous up-regulation in genes affecting membrane permeability and antibiotic efflux [50], all of which would prove advantageous for the bacterium during chronic lung infection. The genetic mutations that bring this about occur as a result of cell envelope stress during unfavourable conditions such as osmotic shock, oxidative stress, magnesium starvation and antimicrobial agents in the inflammatory COPD and CF lung [50,63].
Clinical mucoid sequential clones of P. aeruginosa showed reduced virulence over time, in contrast to the non-mucoid clones which were more virulent in vivo and were associated with reduced clearance [64]. Late non-mucoid isolates of P. aeruginosa have also been obtained from the CF lung; these are suspected revertants of mucoid isolates indicating that the in vivo conditions have changed [65]. The mucoid phenotype switch in P. aeruginosa during chronic infection is associated with poor lung functions, increased anti-EPS antibody titres and poorer outcomes in CF patients [59]. In addition, Huse et al. (2013) demonstrated that non-alginate EPS by P. aeruginosa is just as important as alginate EPS during adaptation to the CF lung [66]. Operons undergoing parallel evolution in vivo enhanced the production of galactose-mannose rich EPS (Psl) in the first 40,000 generations [66,67]. The later chronic clinical isolates produced more Psl than their ancestor strains in 70% of cases in vivo. Psl is required for biofilm formation in mucoid isolates, furthermore, small colony variants overproducing Psl underwent positive selection during chronic colonisation [66].
Zlosnik et al. (2008) showed that mucoid phenotype switches also occurred in sequential isolates from 15 patients with Bcc infection [68], although in contrast to P. aeruginosa, the majority of Bcc isolates showed a mucoid to non-mucoid switch. The non-mucoid Bcc isolates were associated with a more rapid lung function decline than mucoid isolates [69], indicating that this mucoid switch may be associated with increased virulence. In a follow-up study, it was shown that non-mucoid B. cenocepacia isolates were associated with elevated expression of several putative virulence factors. Mutations over time occurred in the manC gene of Bcc [28], which is associated with both EPS and O-antigen synthesis [70]. Mutations were also observed in wpbA and wpbW, which are manC homologues in P. aeruginosa and are associated with reduced O-antigen synthesis in P. aeruginosa over time of colonisation. This suggests that the same forces driving adaptation may also be implicated in niche adaptation within the CF lung in different pathogens [70].
Another Bcc species, B. multivorans, also showed a mucoid to non-mucoid switch, but in contrast to B. cenocepacia and more comparable with P. aeruginosa, showed reduced virulence over time, with the later non-mucoid isolate being less virulent than the clonal mucoid isolate [27]. Reductions in swimming and swarming motility, type IV secretion, haemolysin-type protein secretion and EPS production over time were also observed [27]. In the examination of the structural properties of EPS from Bcc isolates, Herasimenka et al. (2007) found that cepacian was the most common EPS produced by a set of seven clinical isolates including three B. cenocepacia, three B. multivorans and one Bcc member of which taxonomic status had not yet been determined. Only one B. multivorans isolate produced psl, however, no definitive evidence to suggest the presence of cepacian EPS in sputum was noted [71]. It has been suggested however that the production of EPS by B. cenocepacia does confer a selective advantage in vivo by the inhibition of neutrophil chemotaxis and reactive oxygen species [72].
Capsular polysaccharide expression in chronic S. aureus infection was significantly reduced in later isolates relative to earlier isolates, which may be involved in this pathogen’s persistence in vivo [73]. Several chronic infection models of S. aureus indicated altered virulence factor expression over time of colonisation. Proteins associated with host protein interactions (Fbp1) were elevated while the virulence protein haemolysin was reduced indicating enhanced potential to colonise and avoid immune detection [73].
4. Antibiotic Resistance
Antibiotic resistance is a hallmark of chronically colonising pathogens generally and particularly in those associated with CF infections [74,75,76]. Bacteria acquire and utilise different antibiotic resistance mechanisms to protect themselves in vivo including; efflux pumps, porins and altered membrane permeability, enzymatic modification such as β-lactamases which have previously been reviewed [58,77,78]. These resistance mechanisms can be altered during the in vivo course of infection and by exposure to a range of antimicrobial agents, in addition to patient non-compliance [74]. Studies have reported the increased antibiotic resistance of many pulmonary pathogens to a variety of therapeutics over time in vivo [30]. Antibiotic resistance has been associated with modifications in LPS, biofilm formation and QS mutations in P. aeruginosa during chronic infection [12]. OMP’s and in particular multi drug efflux pumps can increase antimicrobial tolerance. These proteins confer resistance to many antibiotics after exposure in vivo which have previously been reviewed [79].
During antibiotic treatment of P. aeruginosa in CF patients, mutations arose in the genes encoding β-lactamases and efflux proteins which confer higher levels of antibiotic resistance. This can potentially be associated with decreased lung function in CF patients over time, but this is not always the case [12,80]. P. aeruginosa isolates from COPD patients also showed a general increased resistance to antibiotics over time. As has been discussed (Section 2.2) antibiotic susceptibility is diverse within single sputum isolates with many morphotypes evident, both sensitive and resistant, and with a wide range of variability among the sensitive isolates [40].
Bacteria from the Bcc are also highly antimicrobially resistant and resistance patterns differ depending on their environmental conditions. As with P. aeruginosa, highly resistant strains of Bcc are isolated from the CF lung more frequently than from non-CF patients. This is most likely as a direct consequence of in vivo adaptation of the microbe to the high levels of administered biocidal agents over time [81]. Early B. cenocepacia clonal isolates were more susceptible to cephalosporins, carbapenem, aminoglycosides, fluoroquinalones and trimethoprim-sulfamethoxazole than subsequent later isolates [82]. In contrast, a late B. multivorans isolate was more susceptible to β-lactam antibiotics, which is not frequently observed in clinical isolates exposed to antibiotics in vivo [27]. It was reported that this may be due to accumulated strain specific mutations and that it is unlikely to be clinically relevant.
Methicillin resistance S. aureus (MRSA) is also a causative agent in nosocomial acquired pneumonia, which has severe implications for individuals with underlying pulmonary disorders such as CF or COPD [83,84]. The small colony variant (SCV) phenotype of S. aureus which is a common phenotypic change during infection in CF patients has been implicated in antibiotic resistance, in particular to aminoglycoside antibiotics, as these SCVs lack an efficient electron transport system, which is required for the antibiotic to enter the cells [12,85]. In addition to this the ability of S. aureus SCV’s to survive within host cells reduces the efficacy of therapeutics against this pathogen [86].
Antibiotic Stress-Effect on the Airway Microbiome
During antibiotic treatment of CF lung infections, it is essential to consider the effect of the biocidal therapy on the co-colonising microorganisms and indeed the lungs’ own microflora. Zemanick et al. [84] compared inflammatory markers and bacterial species in 21 patients with pulmonary exacerbations at early stage (0–3 days) and late stage (7–14 days) of antibiotic treatment. The relative abundance of P. aeruginosa and not anaerobic genera was associated with lower FEV1 values and higher levels of inflammation during antibiotic therapy. In addition, the higher abundance of P. aeruginosa was associated with lower microbial diversity, poorer lung function and increased inflammation, suggesting that the alterations in relative abundance balance of these core anaerobic species, such as Prevotella and Vellionella, may contribute to pulmonary disease [87]. Disruption of the pulmonary microbiome with high levels of biocidal therapies has also been suggested for patients with asthma and COPD [7,88]. Tunney et al. observed that targeted treatment of aerobes had minimal effect on the abundance and diversity of co-colonising anaerobes [89]. More recently, it has been reported that the relative abundance of P. aeruginosa increased in comparison to non-pseudomonal species during antibiotic therapy with anti-pseudomonal antibiotics [90].
5. Motility
A common switch from a highly motile phenotype to a non-flagellated, non-motile variant in P. aeruginosa was reported 20 years ago [91]. This loss in flagella was associated with mutations in flagellar genes including rpoN, vfr, fleQ and fliC. Although the adaptation to a non-motile phenotype during chronic infection was not associated with poorer outcomes, the inability of macrophages to phagocytose non-flagellated P. aeruginosa isolates suggested a potential reason for this phenotypic selection in vivo [29]. More recent studies analysing P. aeruginosa evolution in COPD patients over time of colonisation support this observed reduction in swimming, swarming and twitching motility in chronic infections [30]. Both swimming motility and swarming motility have been associated with biofilm formation [92]. Patankar et al. (2013) demonstrated that this loss in motility caused a reduced inflammasome activation and antibacterial IL-1β host response and could potentially explain how pathogens have evolved strategies to avoid host immune attack [93]. Transcriptional analysis of B. multivorans also showed a decrease in gene transcripts associated with motility and chemotaxis in a late clinical isolate [27]. Conversely numerous B. cenocepacia genes associated with flagella assembly and adhesion were more highly expressed in a later variant when compared to the earlier isolate [31]. In agreement with this a recent study of 551 Bcc isolates from chronic infections, showed that swimming motility was not lost by Bcc, in contrast to P. aeruginosa [94].
6. Morphology Variants
Many bacteria alter their morphology over time of colonisation. In particular, SCVs have a much slower growth rate, which results in smaller colony morphology when grown on routine agar [95]. These SCV’s are often selected for by prolonged antibiotic exposure [65,96], which is common during chronic lung infections. S. aureus SCV’s are associated with persistence in the airways. These slower to grow, antibiotic resistant, non-pigmented colonies also alter their virulence factor expression, for example, reduced expression of α-toxin in S. aureus SCV’s. The high expression of fibronectin binding proteins (FnBPs) in SCV S. aureus promotes their own uptake in host cells which proves beneficial [32]. Once located intracellularly these SCV’s avoid immune activation by reduced host tissue damage and bacterial protease secretions. They are better adapted for intracellular survival in higher numbers and better able to avoid and withstand antimicrobial activity elicited by host cells than their wild type counterparts. The dynamic ability of SCV’s to revert to their wild type phenotype also play a role in chronic infection and aid in the pathogens ability to chronically infect its host [32]. This persistence is potentially related to their greater resistance to host immune defences and antibiotics and their greater ability to adhere to respiratory epithelial cells [97].
In P. aeruginosa, SCVs can also exhibit hyperadherent and autoaggregative behaviour which can favour biofilm formation and persistence in vivo [98,99]. B. multivorans and B. cepacia also express the SCV phenotype which has led to fatal outcomes after bilateral lung transplantation in two patients out of three with CF [100]. The SCV morphotype of these Bcc isolates demonstrated an increased resistance to serum relative to the wild type. Bcc have also been shown to undergo shiny colony variant (SHV), with a switch from a matte colony appearance to SHV which was associated with persistence in mice [101]. This conversion has been associated with reduced virulence and biofilm formation in B. cenocepacia K562, with SHV producing significantly less lung histopathology than the rough K562 strain in a mouse agar bead model of chronic infection [102]. Other pleiotropic effects occur including reduction in motility, biofilm formation, extracellular matrices and siderophores noted [103].
Rugose small colony variants (RSCV) of P. aeruginosa CF isolates have been examined in a study by Starkey et al. (2009) using a combination of transcriptional profiling and Biolog phenotypic analysis. RSCV’s are small wrinkled colonies that have an increased auto-aggregative and biofilm formation capacity. RSCV’s selected for during persistent infection showed increased expression of the pel and psl polysaccharide gene clusters and decreased expression of flagellum and pilus genes [19,33].
7. Quorum Sensing
Prokaryotes generally behave as single cellular organisms in low population densities, however once they sense that the population density has reached a desired level in the formation of biofilms. This QS process allows cells to communicate with each other using small signalling molecules, autoinducers and enables the bacteria to alter the expression of different virulence genes essential for pathogenicity. Mutations in two QS systems LasR and RhlR, which result in loss of QS in clinical P. aeruginosa isolates, were more frequently identified later in CF infection. These QS mutants demonstrated a growth advantage with low levels of amino acids which is particularly relevant in a pulmonary environment and over time of chronic infection in vivo [35,104]. Since virulence factors are generally selected against during chronic infection in vivo, and as QS controls a variety of different virulence factor expression, QS mechanisms could be potentially detrimental to the bacterium during chronic infection [12]. Another advantage in down regulation of QS mechanisms could be attributed to P. aeruginosa QS mutants having increased β-lactamase activity in vitro [35]. In an attempt to examine the social behaviour of P. aeruginosa during chronic infection Jiricny et al., (2014) showed a statistically significant reduction in the 3-oxo-C12-HSL and Pseudomonas Quinalone Signal (PQS) systems [105].
In contrast to P. aeruginosa, McKeon et al. showed that B. cenocepacia QS mutants were far less frequent during chronic infection. Only 1 in 22 patients harboured mutations in both QS Bcc systems, cepR and cciR, which would have an effect on function, the loss in QS function did not appear to confer a competitive advantage for B. cenocepacia in vivo [106]. Recent data showed that a triple knock-out mutant in J2315 of cepl, cciR and Burkholderia diffusible signal factor, had reduced biofilm forming capacities, was more susceptible to antibiotics, and was less virulent in C. elegans than the wild type strain [107], which may explain the lack of QS mutants in chronically colonised CF patients.
S. aureus utilise an accessory gene regulator (agr) QS system which is essential for pathogenesis, biofilm formation and adhesion. S. aureus also utilises a LuxS system in biofilm formation [108]. Interestingly agr mutants are common in chronic CF infection, these mutants are often non-haemolytic with attenuated virulence that cannot lyse blood cells [36].
8. Interactions with Co-Colonising Fungi
In pulmonary diseases including CF and COPD it is accepted that fungal infections including Aspergillus and yeast Candida species are frequently observed and are associated with varied outcomes in the patients infected [109,110]. The effect of co-colonising fungi on pathogenic bacteria cannot be overlooked. In a study of chronically infected CF patients A. fumigatus was identified in 45.7% of patients and significantly C. albicans was isolated from 75.5% of patients [111]. Interestingly an indirect correlation between the richness and diversity of both bacterial and fungal species in the lungs of CF patients and poorer clinical outcomes has been observed, highlighting the role of both commensal and opportunistic pathogens and fungi in pulmonary diseases [112]. The interactions of bacterial and fungal species are evident in the biofilm structures formed in vitro during co-colonisation with P. aeruginosa and A. fumigatus. P. aeruginosa alone formed loosely adherent biofilms in monoculture, however when co-cultured with already pre-established A. fumigatus mycelia, firmly adherent biofilm structures were observed which were comparable with A. fumigatus biofilm structure alone [113]. Mowat et al. subsequently showed diffusible factors produced by P. aeruginosa could inhibit and disrupt biofilm formation of A. fumigatus [114]. Furthermore, the extent of P. aeruginosa induced lung injury in BALB/c mice was shown to be reduced when preceded by airway colonisation by C. albicans, an effect that was reversed by treatment with the antifungal, capsofungin. This highlights C. albicans could modulate the virulence of P. aeruginosa in an in vivo model again highlighting the complex relationship between these species in vivo [115].
9. Methodologies Exploited in the Study of Adaptation
9.1. Microarrays in the Study of Microbial Adaptation and Pathogenesis
In the last few decades major advances have been made in the study of single genes to whole genome sequencing in pathogens. A number of genomic tools have been used to determine virulence factors involved in pathogenesis. With increasing numbers of bacterial genomes published, comparative genomics and expression analysis has come to the fore. In particular, microarrays have been powerful tools in the determination of alterations in gene expression during the course of clinical infection or comparison of virulence determinants in different lung pathogens. The use of cDNA microarrays allows the rapid elucidation of gene expression divergence among sequential clinical isolates during the course of infection. The analysis of genes involved in virulence can be performed on numerous pathogens simultaneously, which is advantageous when dealing with large numbers of sequential isolates. The evolution of bacterial genetics at the DNA level can also be studied using these robotic chip-based systems [116]. Microarrays and independent component analysis proved useful in determining that P. aeruginosa utilises multiple patient specific and parallel adaptations during chronic CF infection. The common parallel adaptations revealed that earlier isolates had higher levels of type three secretion systems and exoenzyme activities than later isolates, whereas the later isolates generally shared antimicrobial peptide resistance genes, alginate production genes and QS genes [117]. Other applications of this technology in elucidating certain adaptations that pulmonary pathogens undergo during chronic infection have already been discussed in this review.
The limitation associated with this technology is that robotics and array fabrication can be variable and data analysis complex. Careful designing of an experiment, improved system components and quality control steps at each step of the process can eliminate these variables. Importantly microarray analysis on DNA and mRNA transcripts levels does not fully depict the protein expression levels within a cell and the gene function in microbes, with protein translations and modification all impacting on the final functional molecules in the cells. Nevertheless this technology has provided great insight into the ability of microbes to diversify during chronic lung infections [118].
9.2. Proteomics in the Study of Microbial Adaptation and Pathogenesis
The utilisation of proteomic approaches in bacterial evolution has been discussed throughout this review. Madeira et al. utilised a 2D Difference gel electrophoresis (2D DIGE) to determine evolution strategies employed by three sequential CF B. cenocepacia isolates from a total of 11 isolates. These three isolates included the initial isolate and two later isolates (one following an exacerbation and one IST4134 retrieved 3.5 years later immediately prior to patient death). The proteomic analysis correlated well with alterations in phenotype. The two later isolates were more capable of invading and disrupting 16HBE14o- and CFBE41o-cells in vitro and were also better able to cope in iron limited conditions which collectively shows a beneficial adaptive strategy employed by the pathogen to survive over time in the CF lung. A metabolic reprogramming was also speculated [21]. In an earlier study by Chung and Speert (2006) a combination of 2D gel and MALDI-ToF mass spectrometry were utilised to determine the proteins necessary for B. cenocepacia survival in a murine host, they noted that alkaline hydroperoxide reductase subunit C (ahpC) protein spot was lost in the persistent isolate. AhpC expression has been associated with resistance to oxidative stress. An increase in flagellar genes was also observed, which may be associated with bacterial persistence [119]. Gel based systems provide a relatively straight forward method to study proteomics; however with the development of MS to complement these approaches gel based proteomics are getting less scope recently. Limitations associated with gel based proteomics include poor representation of low abundant proteins, difficulty to resolve and separate highly acidic/basic/hydrophobic and large proteins. In addition to this the potential for multiple proteins to be present in a single spot can result in inaccurate comparisons [120]. LC/MS proteomic platforms avoid some of these issues and have been successful in examining non-protein QS signalling molecules, for example [105].
Limitations to this type of analysis include; total accurate quantification of a particular protein is only possible when the peptides are exclusively derived from a particular protein. In addition to this quantification of post translational modifications is still developing [121]. Nevertheless this platform has proved extremely valuable in determining pathogen adaptation during chronic lung infection.
10. Conclusions
Chronic lung infections are associated with increased morbidity and mortality for individuals with underlying pulmonary disorders, particularly for CF and COPD patients. The pathogenesis and adaptation mechanisms utilised by Gram positive and Gram negative bacteria during chronic lung infection involves numerous complex and diverse virulence factors. These determinants include surface components that are in direct contact with the host such as adhesins and outer-membrane proteins and pyrogenic LPS involved in the initial colonisation step. Antibiotic resistance, biofilm formation, EPS production, hypermutability and colony variants, which are frequently reported during chronic infection can be altered through the clinical course of infection to assist in the survival and persistence of these pathogenic pulmonary pathogens. It is becoming apparent that while P. aeruginosa and other species, such as B. multivorans reduce their virulence over time, B. cenocepacia, contrary to expectations, appears to increase its virulence (for example, non-mucoid phenotype, flagella). The expression and repression of these virulence factors during chronic infection involves an intricate regulation system unique to a bacterial species, isolate or cell that ultimately provides the pathogen with a competitive edge in vivo. These systems include QS systems and mutations in genes responsible for various virulence factor regulations, for example MucA. The frequency of mutations in mucA and QS in P. aeruginosa would suggest that loss of function of these virulence regulatory genes is advantageous in vivo and selective pressures within the host select for these mutants.
The evolution of bacteria is extremely complex and cannot be explained by a single evolutionary trajectory. It is more plausible that numerous simultaneous and parallel mutations select for pathogenic communities that are better suited to their niche. The host lung environment, co-infecting microbes and cell specific factors all contribute to the diversity of these pathogens, which makes extrapolation and comparison of these opportunists difficult. Nevertheless these pathogens have one common goal, which is to survive within the host lungs by evasion of host defences and antimicrobial therapies. Further studies in this area using a combination of molecular, proteomic and phenotypic studies will contribute to the knowledge of bacterial pathogenesis and evolution and provide potential therapeutic targets in eradicating these frequently antibiotic resistant pathogens.
Author Contributions
L. C. and S. McC. jointly wrote the review.
Acknowledgements
L. C. is funded by the Irish Research Council. S. McC. is a member of the EU COST Action the EU COST Action BM1003: Microbial cell surface determinants of virulence as targets for new therapeutics in cystic fibrosis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eisele, N.A.; Anderson, D.M. Host defense and the airway epithelium: Frontline responses that protect against bacterial invasion and pneumonia. J. Pathog. 2011, 2011, 249802. [Google Scholar] [PubMed]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care. Med. 2011, 184, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Zakharkina, T.; Heinzel, E.; Koczulla, R.A.; Greulich, T.; Rentz, K.; Pauling, J.K.; Baumbach, J.; Herrmann, M.; Grunewald, C.; Dienemann, H. Analysis of the airway microbiota of healthy individuals and patients with chronic obstructive pulmonary disease by t-rflp and clone sequencing. PLoS One 2013, 8, e68302. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Prescott, H.C.; Martinez, F.J.; Curtis, J.L.; Lama, V.N.; Huffnagle, G.B. Cell-associated bacteria in the human lung microbiome. Microbiome 2014, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.M.; Young, V.B.; Huffnagle, G.B. The microbiome of the lung. Transl. Res. 2012, 160, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Spurzem, J.R.; Rennard, S.I. Pathogenesis of COPD. Semin. Respir. Crit. Care. Med. 2005, 26, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L. Disordered microbial communities in asthmatic airways. PLoS One 2010, 5, e8578. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.K. Mucus, phlegm, and sputum in cystic fibrosis. Respir Care 2009, 54, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.J.; Allgaier, M.; Taylor, B.; Baek, M.S.; Huang, Y.J.; Daly, R.A.; Karaoz, U.; Andersen, G.L.; Brown, R.; Fujimura, K.E. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One 2010, 5, e11044. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, M.; McClean, S. Bacterial host interactions in cystic fibrosis. Curr. Opin. Microbiol. 2012, 15, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.R.; Jain, M.; Bar-Meir, M.; McColley, S.A. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin. Microbiol. Rev. 2011, 24, 29–70. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.F.; Brauer, A.L.; Grant, B.J.; Sethi, S. Moraxella catarrhalis in chronic obstructive pulmonary disease: Burden of disease and immune response. Am. J. Respir. Crit. Care. Med. 2005, 172, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.F.; Brauer, A.L.; Eschberger, K.; Lobbins, P.; Grove, L.; Cai, X.; Sethi, S. Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 2008, 177, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Cornforth, D.M.; Mideo, N. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends. Microbiol. 2012, 20, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wang, A.H.; Jennings, M.P. Discovery of virulence factors of pathogenic bacteria. Curr. Opin. Chem. Biol. 2008, 12, 93–101. [Google Scholar] [CrossRef] [PubMed]
- McClean, S. Eight stranded β-barrel and related outer membrane proteins: Role in Bacterial pathogenesis. Protein Pept. Lett. 2012, 19, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Dennehy, R.; McClean, S. Immunoproteomics: The key to discovery of new vaccine antigens against bacterial respiratory infections. Curr. Protein Pept. Sci. 2012, 13, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D'Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar] [CrossRef] [PubMed]
- McAdam, P.R.; Holmes, A.; Templeton, K.E.; Fitzgerald, J.R. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS One 2011, 6, e24301. [Google Scholar] [CrossRef]
- Madeira, A.; dos Santos, S.C.; Santos, P.M.; Coutinho, C.P.; Tyrrell, J.; McClean, S.; Callaghan, M.; Sa-Correia, I. Proteomic profiling of Burkholderia cenocepacia clonal isolates with different virulence potential retrieved from a cystic fibrosis patient during chronic lung infection. PLoS One 2013. [Google Scholar] [CrossRef]
- Marvig, R.L.; Damkiaer, S.; Khademi, S.M.; Markussen, T.M.; Molin, S.; Jelsbak, L. Within-host evolution of Pseudomonas aeruginosa reveals adaptation toward iron acquisition from hemoglobin. MBio 2014, 5, e00966-14. [Google Scholar] [CrossRef]
- Nakamura, S.; Shchepetov, M.; Dalia, A.B.; Clark, S.E.; Murphy, T.F.; Sethi, S.; Gilsdorf, J.R.; Smith, A.L.; Weiser, J.N. Molecular basis of increased serum resistance among pulmonary isolates of non-typeable Haemophilus influenzae. PLoS Pathog. 2011, 7, e1001247. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Mutharia, L.M.; Chan, L.; Darveau, R.P.; Speert, D.P.; Pier, G.B. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: A class of serum-sensitive, nontypable strains deficient in lipopolysaccharide O side chains. Infect. Immun. 1983, 42, 170–177. [Google Scholar] [PubMed]
- Madeira, A.; Santos, P.M.; Coutinho, C.P.; Pinto-de-Oliveira, A.; Sa-Correia, I. Quantitative proteomics (2-d dige) reveals molecular strategies employed by Burkholderia cenocepacia to adapt to the airways of cystic fibrosis patients under antimicrobial therapy. Proteomics 2011, 11, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.W.; Schurr, M.J.; Mudd, M.H.; Govan, J.R.; Holloway, B.W.; Deretic, V. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 1993, 90, 8377–8381. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.N.; Ferreira, A.S.; Becker, J.D.; Zlosnik, J.E.; Speert, D.P.; He, J.; Mil-Homens, D.; Moreira, L.M. Mucoid morphotype variation of Burkholderia multivorans during chronic cystic fibrosis lung infection is correlated with changes in metabolism, motility, biofilm formation and virulence. Microbiology 2011, 157, 3124–3137. [Google Scholar] [CrossRef]
- Zlosnik, J.E.; Speert, D.P. The role of mucoidy in virulence of bacteria from the Burkholderia cepacia complex: A systematic proteomic and transcriptomic analysis. J. Infect. Dis. 2010, 202, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam, E.; Campbell, M.E.; Speert, D.P. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 1994, 62, 596–605. [Google Scholar] [PubMed]
- Martinez-Solano, L.; Macia, M.D.; Fajardo, A.; Oliver, A.; Martinez, J.L. Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin. Infect. Dis. 2008, 47, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Mira, N.P.; Madeira, A.; Moreira, A.S.; Coutinho, C.P.; Sa-Correia, I. Genomic expression analysis reveals strategies of Burkholderia cenocepacia to adapt to cystic fibrosis patients' airways and antimicrobial therapy. PLoS One 2011, 6, e28831. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Heitmann, V.; Hussain, M.; Viemann, D.; Roth, J.; von Eiff, C.; Peters, G.; Becker, K.; Loffler, B. Staphylococcus aureus small-colony variants are adapted phenotypes for intracellular persistence. J. Infect. Dis. 2010, 202, 1031–1040. [Google Scholar] [CrossRef]
- Starkey, M.; Hickman, J.H.; Ma, L.; Zhang, N.; De Long, S.; Hinz, A.; Palacios, S.; Manoil, C.; Kirisits, M.J.; Starner, T.D. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 2009, 191, 3492–3503. [Google Scholar] [CrossRef]
- Subramoni, S.; Nguyen, D.T.; Sokol, P.A. Burkholderia cenocepacia shvr-regulated genes that influence colony morphology, biofilm formation, and virulence. Infect. Immun. 2011, 79, 2984–2997. [Google Scholar] [CrossRef] [PubMed]
- D'Argenio, D.A.; Wu, M.; Hoffman, L.R.; Kulasekara, H.D.; Deziel, E.; Smith, E.E.; Nguyen, H.; Ernst, R.K.; Larson Freeman, T.J.; Spencer, D.H. Growth phenotypes of Pseudomonas aeruginosa lasr mutants adapted to the airways of cystic fibrosis patients. Mol. Microbiol. 2007, 64, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Goerke, C.; Wolz, C. Regulatory and genomic plasticity of Staphylococcus aureus during persistent colonization and infection. Int J. Med. Microbiol. 2004, 294, 195–202. [Google Scholar] [CrossRef]
- Schmidt, H.; Hensel, M. Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 2004, 17, 14–56. [Google Scholar] [CrossRef] [PubMed]
- Pohl, K.; Francois, P.; Stenz, L.; Schlink, F.; Geiger, T.; Herbert, S.; Goerke, C.; Schrenzel, J.; Wolz, C. Cody in Staphylococcus aureus: A regulatory link between metabolism and virulence gene expression. J. Bacteriol 2009, 191, 2953–2963. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, J.L.; Mowat, E.; Ledson, M.J.; Walshaw, M.J.; Winstanley, C. Fluctuations in phenotypes and genotypes within populations of Pseudomonas aeruginosa in the cystic fibrosis lung during pulmonary exacerbations. J. Med. Microbiol 2010, 59, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Workentine, M.L.; Sibley, C.D.; Glezerson, B.; Purighalla, S.; Norgaard-Gron, J.C.; Parkins, M.D.; Rabin, H.R.; Surette, M.G. Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One 2013, 8, e60225. [Google Scholar] [CrossRef] [PubMed]
- Rakhimova, E.; Wiehlmann, L.; Brauer, A.L.; Sethi, S.; Murphy, T.F.; Tummler, B. Pseudomonas aeruginosa population biology in chronic obstructive pulmonary disease. J. Infect. Dis. 2009, 200, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.S.; Coutinho, C.P.; Azevedo, P.; Lito, L.; Melo-Cristino, J.; Sa-Correia, I. Burkholderia dolosa phenotypic variation during the decline in lung function of a cystic fibrosis patient during 5.5 years of chronic colonization. J. Med. Microbiol. 2014, 63, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, T.D.; Flett, K.B.; Yelin, I.; Martin, T.R.; McAdam, A.J.; Priebe, G.P.; Kishony, R. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat. Genet. 2014, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Singh, P.K. Evolving stealth: Genetic adaptation of Pseudomonas aeruginosa during cystic fibrosis infections. Proc. Natl. Acad. Sci. USA 2006, 103, 8305–8306. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A. Mutators in cystic fibrosis chronic lung infection: Prevalence, mechanisms, and consequences for antimicrobial therapy. Int. J. Med. Microbiol. 2010, 300, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rojas, A.; Rodriguez-Beltran, J.; Couce, A.; Blazquez, J. Antibiotics and antibiotic resistance: A bitter fight against evolution. Int. J. Med. Microbiol. 2013, 303, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.E., Jr.; Burns, J.L.; Smith, A.L. Hypermutable haemophilus influenzae with mutations in mutS are found in cystic fibrosis sputum. Microbiology 2004, 150, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Hogardt, M.; Hoboth, C.; Schmoldt, S.; Henke, C.; Bader, L.; Heesemann, J. Stage-specific adaptation of hypermutable Pseudomonas aeruginosa isolates during chronic pulmonary infection in patients with cystic fibrosis. J. Infect. Dis. 2007, 195, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jelsbak, L.; Marvig, R.; Damkiaer, S.; Workman, C.; Rau, M.; Hansen, S.; Folkesson, A.; Johansen, H.; Ciofu, O. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. USA 2011, 108, 7481–7486. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Hoiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Microbiol. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Mossialos, D.; Amoutzias, G.D. Role of siderophores in cystic fibrosis pathogenesis: Foes or friends? Int. J. Med. Microbiol. 2009, 299, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.B.; Majumdar, S.; Hani, E.; Sokol, P.A. Importance of the ornibactin and pyochelin siderophore transport systems in Burkholderia cenocepacia lung infections. Infect. Immun. 2004, 72, 2850–2857. [Google Scholar] [CrossRef] [PubMed]
- Cobessi, D.; Celia, H.; Folschweiller, N.; Schalk, I.J.; Abdallah, M.A.; Pattus, F. The crystal structure of the pyoverdine outer membrane receptor FpvA from Pseudomonas aeruginosa at 3.6 angstroms resolution. J. Mol. Biol. 2005, 347, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Kanoh, S. Involvement of central action of lipopolysaccharide in pyrogen fever. Jpn. J. Pharmacol 1984, 36, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via tlr4/myd88 and the cgmp-dependent protein kinase pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H.; Libby, P.; Freudenberg, M.; Krauss, J.H.; Weckesser, J.; Mayer, H. Cytokine induction by lipopolysaccharide (LPS) corresponds to lethal toxicity and is inhibited by nontoxic rhodobacter capsulatus LPS. Infect. Immun. 1990, 58, 3743–3750. [Google Scholar] [PubMed]
- Jeyaseelan, S.; Chu, H.W.; Young, S.K.; Freeman, M.W.; Worthen, G.S. Distinct roles of pattern recognition receptors cd14 and toll-like receptor 4 in acute lung injury. Infect. Immun. 2005, 73, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Hunyadi, A.; Amaral, L. Mechanisms of resistance in bacteria: An evolutionary approach. Open. Microbiol. J. 2013, 7, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Govan, J.R.; Deretic, V. Microbial pathogenesis in cystic fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 1996, 60, 539–574. [Google Scholar] [PubMed]
- Linker, A.; Jones, R.S. A new polysaccharide resembling alginic acid isolated from pseudomonads. J. Biol. Chem. 1966, 241, 3845–3851. [Google Scholar] [PubMed]
- Schurr, M.J.; Yu, H.; Martinez-Salazar, J.M.; Boucher, J.C.; Deretic, V. Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J. Bacteriol. 1996, 178, 4997–5004. [Google Scholar] [PubMed]
- Mathee, K.; McPherson, C.J.; Ohman, D.E. Posttranslational control of the algT (algU)-encoded sigma22 for expression of the alginate regulon in Pseudomonas aeruginosa and localization of its antagonist proteins MucA and MucB (algN). J. Bacteriol. 1997, 179, 3711–3720. [Google Scholar] [PubMed]
- Schurr, M.J.; Deretic, V. Microbial pathogenesis in cystic fibrosis: Co-ordinate regulation of heat-shock response and conversion to mucoidy in Pseudomonas aeruginosa. Mol. Microbiol. 1997, 24, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; van Gennip, M.; Bjarnsholt, T.; Jensen, P.O.; Lee, B.; Hougen, H.P.; Calum, H.; Ciofu, O.; Givskov, M.; Molin, S. Novel experimental Pseudomonas aeruginosa lung infection model mimicking long-term host-pathogen interactions in cystic fibrosis. APMIS 2009, 117, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Hogardt, M.; Heesemann, J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. 2010, 300, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Huse, H.K.; Kwon, T.; Zlosnik, J.E.; Speert, D.P.; Marcotte, E.M.; Whiteley, M. Pseudomonas aeruginosa enhances production of a non-alginate exopolysaccharide during long-term colonization of the cystic fibrosis lung. PLoS One 2013, 8, e82621. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lu, H.; Sprinkle, A.; Parsek, M.R.; Wozniak, D.J. Pseudomonas aeruginosa psl is a galactose- and mannose-rich exopolysaccharide. J. Bacteriol. 2007, 189, 8353–8356. [Google Scholar] [CrossRef] [PubMed]
- Zlosnik, J.E.; Hird, T.J.; Fraenkel, M.C.; Moreira, L.M.; Henry, D.A.; Speert, D.P. Differential mucoid exopolysaccharide production by members of the Burkholderia cepacia complex. J. Clin. Microbiol. 2008, 46, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Zlosnik, J.E.; Costa, P.S.; Brant, R.; Mori, P.Y.; Hird, T.J.; Fraenkel, M.C.; Wilcox, P.G.; Davidson, A.G.; Speert, D.P. Mucoid and nonmucoid Burkholderia cepacia complex bacteria in cystic fibrosis infections. Am. J. Respir. Crit. Care. Med. 2010, in press. [Google Scholar]
- Traverse, C.C.; Mayo-Smith, L.M.; Poltak, S.R.; Cooper, V.S. Tangled bank of experimentally evolved Burkholderia biofilms reflects selection during chronic infections. Proc. Natl. Acad. Sci. USA 2013, 110, E250–E259. [Google Scholar] [CrossRef] [PubMed]
- Herasimenka, Y.; Cescutti, P.; Impallomeni, G.; Campana, S.; Taccetti, G.; Ravenni, N.; Zanetti, F.; Rizzo, R. Exopolysaccharides produced by clinical strains belonging to the Burkholderia cepacia complex. J. Cyst. Fibros. 2007, 6, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Bylund, J.; Burgess, L.A.; Cescutti, P.; Ernst, R.K.; Speert, D.P. Exopolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge reactive oxygen species. J. Biol. Chem. 2006, 281, 2526–2532. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Medina, E.; Hussain, M.; Volker, W.; Heitmann, V.; Niemann, S.; Holzinger, D.; Roth, J.; Proctor, R.A.; Becker, K. Staphylococcus aureus phenotype switching: An effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 2011, 3, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Johnston, S.L. The role of antibiotics in asthma. Int. J. Antimicrob. Ag. 2007, 29, 485–493. [Google Scholar] [CrossRef]
- Khanbabaee, G.; Akbarizadeh, M.; Sayyari, A.; Ashayeri-Panah, M.; Abdollahgorji, F.; Sheibani, K.; Rezaei, N. A survey on pulmonary pathogens and their antibiotic susceptibility among cystic fibrosis patients. Braz. J. Infect. Dis. 2012, 16, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Tarsia, P.; Pappalettera, M.; Saporiti, M.; Aliberti, S. Antibiotic therapy and prophylaxis in COPD. Resp Med: COPD Update 2007, 2, 124–132. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Alanis, A.J. Resistance to antibiotics: Are we in the post-antibiotic era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.S. Efflux systems in bacterial pathogens: An opportunity for therapeutic intervention? An industry view. Biochem. Pharmacol. 2006, 71, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.L.; Konstan, M.W.; Yegin, A.; Rasouliyan, L.; Trzaskoma, B.; Morgan, W.J.; Regelmann, W.; Scientific Advisory Group, I.; Coordinators of the Epidemiologic Study of Cystic Fibrosis. Multiple antibiotic-resistant Pseudomonas aeruginosa and lung function decline in patients with cystic fibrosis. J. Cyst. Fibros. 2012, 11, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, Y.; Tabibi, S.; Alba, L.; Garber, E.; Saiman, L. Antimicrobial susceptibility and synergy studies of Burkholderia cepacia complex isolated from patients with cystic fibrosis. Antimicrob. Ag. Chemother. 2007, 51, 1085–1088. [Google Scholar] [CrossRef]
- Coutinho, C.P.; de Carvalho, C.C.; Madeira, A.; Pinto-de-Oliveira, A.; Sa-Correia, I. Burkholderia cenocepacia phenotypic clonal variation during a 3.5-year colonization in the lungs of a cystic fibrosis patient. Infect. Immun. 2011, 79, 2950–2960. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Pletz, M.W. Antimicrobial treatment of nosocomial meticillin-resistant Staphylococcus aureus (MRSA) pneumonia: Current and future options. Int. J. Antimicrob. Ag. 2010, 36, 391–400. [Google Scholar] [CrossRef]
- Goss, C.H.; Muhlebach, M.S. Review: Staphylococcus aureus and MRSA in cystic fibrosis. J. Cyst. Fibros. 2011, 10, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Balwit, J.M.; van Langevelde, P.; Vann, J.M.; Proctor, R.A. Gentamicin-resistant menadione and hemin auxotrophic Staphylococcus aureus persist within cultured endothelial cells. J. Infect. Dis. 1994, 170, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Von Eiff, C.; Becker, K.; Metze, D.; Lubritz, G.; Hockmann, J.; Schwarz, T.; Peters, G. Intracellular persistence of Staphylococcus aureus small-colony variants within keratinocytes: A cause for antibiotic treatment failure in a patient with darier’s disease. Clin. Infect. Dis. 2001, 32, 1643–1647. [Google Scholar]
- Zemanick, E.T.; Harris, J.K.; Wagner, B.D.; Robertson, C.E.; Sagel, S.D.; Stevens, M.J.; Accurso, F.J.; Laguna, T.A. Inflammation and airway microbiota during cystic fibrosis pulmonary exacerbations. PLoS One 2013, 8, e62917. [Google Scholar] [CrossRef] [PubMed]
- Erb-Downward, J.R.; Thompson, D.L.; Han, M.K.; Freeman, C.M.; McCloskey, L.; Schmidt, L.A.; Young, V.B.; Toews, G.B.; Curtis, J.L.; Sundaram, B. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 2011, 6, e16384. [Google Scholar] [CrossRef] [PubMed]
- Tunney, M.M.; Klem, E.R.; Fodor, A.A.; Gilpin, D.F.; Moriarty, T.F.; McGrath, S.J.; Muhlebach, M.S.; Boucher, R.C.; Cardwell, C.; Doering, G. Use of culture and molecular analysis to determine the effect of antibiotic treatment on microbial community diversity and abundance during exacerbation in patients with cystic fibrosis. Thorax 2011, 66, 579–584. [Google Scholar]
- Daniels, T.W.; Rogers, G.B.; Stressmann, F.A.; van der Gast, C.J.; Bruce, K.D.; Jones, G.R.; Connett, G.J.; Legg, J.P.; Carroll, M.P. Impact of antibiotic treatment for pulmonary exacerbations on bacterial diversity in cystic fibrosis. J. Cyst. Fibros. 2013, 12, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam, E.; Campbell, M.E.; Speert, D.P. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 1994, 62, 596–605. [Google Scholar] [PubMed]
- Kirov, S.M. Bacteria that express lateral flagella enable dissection of the multifunctional roles of flagella in pathogenesis. FEMS Microbiol. Lett. 2003, 224, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Patankar, Y.R.; Lovewell, R.R.; Poynter, M.E.; Jyot, J.; Kazmierczak, B.I.; Berwin, B. Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect. Immun. 2013, 81, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Zlosnik, J.E.; Mori, P.Y.; To, D.; Leung, J.; Hird, T.J.; Speert, D.P. Swimming motility in a longitudinal collection of clinical isolates of Burkholderia cepacia complex bacteria from people with cystic fibrosis. PLoS One 2014, 9, e106428. [Google Scholar] [CrossRef] [PubMed]
- Chuard, C.; Vaudaux, P.E.; Proctor, R.A.; Lew, D.P. Decreased susceptibility to antibiotic killing of a stable small colony variant of Staphylococcus aureus in fluid phase and on fibronectin-coated surfaces. J. Antimicrob. Chemother. 1997, 39, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Schaaff, F.; Bierbaum, G.; Baumert, N.; Bartmann, P.; Sahl, H.G. Mutations are involved in emergence of aminoglycoside-induced small colony variants of Staphylococcus aureus. Int. J. Med. Microbiol. 2003, 293, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Denton, S.C.M. Staphylococcus aureus and MRSA; Karger: Basel, Switzerland, 2006; Volume 34, pp. 153–159. [Google Scholar]
- Von Gotz, F.; Haussler, S.; Jordan, D.; Saravanamuthu, S.S.; Wehmhoner, D.; Strussmann, A.; Lauber, J.; Attree, I.; Buer, J.; Tummler, B. Expression analysis of a highly adherent and cytotoxic small colony variant of Pseudomonas aeruginosa isolated from a lung of a patient with cystic fibrosis. J. Bacteriol. 2004, 186, 3837–3847. [Google Scholar]
- Kirisits, M.J.; Prost, L.; Starkey, M.; Parsek, M.R. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 2005, 71, 4809–4821. [Google Scholar] [CrossRef] [PubMed]
- Haussler, S.; Lehmann, C.; Breselge, C.; Rohde, M.; Classen, M.; Tummler, B.; Vandamme, P.; Steinmetz, I. Fatal outcome of lung transplantation in cystic fibrosis patients due to small-colony variants of the Burkholderia cepacia complex. Eur. J. Clin. Microbiol. 2003, 22, 249–253. [Google Scholar] [CrossRef]
- Chung, J.W.; Altman, E.; Beveridge, T.J.; Speert, D.P. Colonial morphology of Burkholderia cepacia complex genomovar iii: Implications in exopolysaccharide production, pilus expression, and persistence in the mouse. Infect. Immun. 2003, 71, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Bernier, S.P.; Nguyen, D.T.; Sokol, P.A. A lysr-type transcriptional regulator in Burkholderia cenocepacia influences colony morphology and virulence. Infect. Immun. 2008, 76, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Loutet, S.A.; Valvano, M.A. A decade of Burkholderia cenocepacia virulence determinant research. Infect. Immun. 2010, 78, 4088–4100. [Google Scholar] [CrossRef] [PubMed]
- Oberhardt, M.A.; Goldberg, J.B.; Hogardt, M.; Papin, J.A. Metabolic network analysis of Pseudomonas aeruginosa during chronic cystic fibrosis lung infection. J. Bacteriol. 2010, 192, 5534–5548. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, N.; Molin, S.; Foster, K.; Diggle, S.P.; Scanlan, P.D.; Ghoul, M.; Johansen, H.K.; Santorelli, L.A.; Popat, R.; West, S.A. Loss of social behaviours in populations of Pseudomonas aeruginosa infecting lungs of patients with cystic fibrosis. PLoS One 2014, 9, e83124. [Google Scholar] [CrossRef] [PubMed]
- McKeon, S.A.; Nguyen, D.T.; Viteri, D.F.; Zlosnik, J.E.; Sokol, P.A. Functional quorum sensing systems are maintained during chronic Burkholderia cepacia complex infections in patients with cystic fibrosis. J. Infect. Dis. 2011, 203, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Udine, C.; Brackman, G.; Bazzini, S.; Buroni, S.; Van Acker, H.; Pasca, M.R.; Riccardi, G.; Coenye, T. Phenotypic and genotypic characterisation of Burkholderia cenocepacia J2315 mutants affected in homoserine lactone and diffusible signal factor-based quorum sensing systems suggests interplay between both types of systems. PLoS One 2013, 8, e55112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, K.F.; Vuong, C.; Otto, M. Staphylococcus quorum sensing in biofilm formation and infection. Int. J. Med. Microbiol. 2006, 296, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Maiz, L.; Cuevas, M.; Lamas, A.; Sousa, A.; Quirce, S.; Suarez, L. Aspergillus fumigatus and Candida albicans in cystic fibrosis: Clinical significance and specific immune response involving serum immunoglobulins G, A, and M]. Arch. Bronconeumol. 2008, 44, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Ader, F.; Bienvenu, A.L.; Rammaert, B.; Nseir, S. Management of invasive aspergillosis in patients with COPD: Rational use of voriconazole. Int. J. Chron. Obstruct. Pulmon. Dis. 2009, 4, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Bakare, N.; Rickerts, V.; Bargon, J.; Just-Nubling, G. Prevalence of Aspergillus fumigatus and other fungal species in the sputum of adult patients with cystic fibrosis. Mycoses 2003, 46, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Delhaes, L.; Monchy, S.; Frealle, E.; Hubans, C.; Salleron, J.; Leroy, S.; Prevotat, A.; Wallet, F.; Wallaert, B.; Dei-Cas, E. The airway microbiota in cystic fibrosis: A complex fungal and bacterial community—implications for therapeutic management. PLoS One 2012, 7, e36313. [Google Scholar] [CrossRef] [PubMed]
- Manavathu, E.K.; Vager, D.L.; Vazquez, J.A. Development and antimicrobial susceptibility studies of in vitro monomicrobial and polymicrobial biofilm models with Aspergillus fumigatus and Pseudomonas aeruginosa. BMC Microbiol. 2014, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Mowat, E.; Rajendran, R.; Williams, C.; McCulloch, E.; Jones, B.; Lang, S.; Ramage, G. Pseudomonas aeruginosa and their small diffusible extracellular molecules inhibit Aspergillus fumigatus biofilm formation. FEMS Microbiol. Lett. 2010, 313, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Ader, F.; Jawhara, S.; Nseir, S.; Kipnis, E.; Faure, K.; Vuotto, F.; Chemani, C.; Sendid, B.; Poulain, D.; Guery, B. Short term Candida albicans colonization reduces Pseudomonas aeruginosa-related lung injury and bacterial burden in a murine model. Crit. Care. 2011, 15, R150. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.R.; Musser, J.M. Evolutionary genomics of pathogenic bacteria. Trends Microbiol. 2001, 9, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rau, M.H.; Yang, L.; Hoiby, N.; Molin, S.; Jelsbak, L. Bacterial adaptation during chronic infection revealed by independent component analysis of transcriptomic data. BMC Microbiol. 2011, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Jaluria, P.; Konstantopoulos, K.; Betenbaugh, M.; Shiloach, J. A perspective on microarrays: Current applications, pitfalls, and potential uses. Microb. Cell. Fact. 2007, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Speert, D.P. Proteomic identification and characterization of bacterial factors associated with Burkholderia cenocepacia survival in a murine host. Microbiology 2007, 153, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.; Dumas-Gaudot, E.; Renaut, J.; Sergeant, K. Gel-based and gel-free quantitative proteomics approaches at a glance. Int J. Plant Genomics 2012, 2012, 494572. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, T.; Qian, W.J.; Petyuk, V.A.; Smith, R.D. Liquid chromatography-mass spectrometry-based quantitative proteomics. J. Biol. Chem. 2011, 286, 25443–25449. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cullen, L.; McClean, S. Bacterial Adaptation during Chronic Respiratory Infections. Pathogens 2015, 4, 66-89. https://doi.org/10.3390/pathogens4010066
AMA Style
Cullen L, McClean S. Bacterial Adaptation during Chronic Respiratory Infections. Pathogens. 2015; 4(1):66-89. https://doi.org/10.3390/pathogens4010066
Chicago/Turabian StyleCullen, Louise, and Siobhán McClean. 2015. "Bacterial Adaptation during Chronic Respiratory Infections" Pathogens 4, no. 1: 66-89. https://doi.org/10.3390/pathogens4010066