Article Text

Abstract
Objective Experimental and human data suggest that tumour necrosis factor (TNF) blockade may affect B cell responses, in particular the induction of T cell-dependent (TD) humoral immunity. This study aimed to assess this hypothesis directly in patients with arthritis by analysing longitudinally the effect of TNF blockade on B cell activation and the maturation of humoral responses against TD and T cell-independent vaccines.
Materials and methods Peripheral blood samples were obtained from 56 spondyloarthritis patients before and after treatment with either non-steroidal anti-inflammatory drug (NSAID) alone or TNF blockers and analysed for B cell activation, plasma cell differentiation, germinal centre versus extra-follicular B cell maturation, and somatic hypermutation. Vaccine responses to hepatitis B and Streptococcus pneumoniae were measured by ELISA.
Results TNF blockade augmented B cell activation as reflected by the expression of early activation markers, CD40, and costimulatory molecules, without affecting differentiation towards plasmablasts. This was associated with a specific increase of the unswitched fraction of circulating memory B cells and a decreased level of somatic hypermutation in anti-TNF treated patients, indicating an impairment of the germinal centre-dependent B cell maturation. In agreement with these findings, TNF blockade profoundly suppressed the response to the TD vaccination against hepatitis B, whereas the T cell-independent response against pneumococcal polysaccharides was only modestly affected.
Conclusions These data indicate that TNF blockade severely impedes the induction of primary TD humoral responses, probably by interfering with the germinal centre reaction.
- B cells
- Vaccination
- Anti-TNF
- Autoantibodies
- T Cells
Statistics from Altmetric.com
Introduction
Anti- tumour necrosis factor (TNF) treatment is a potent treatment for rheumatoid arthritis (RA), spondyloarthritis (SpA), Crohn's disease and psoriasis. Therapeutic blockade of this cytokine interferes at multiple levels with inflammatory cascades1 but has also unpredicted effects such as interference with nociceptive brain activity2 and the Fc-mediated induction of anti-inflammatory macrophages by anti-TNF antibodies.3 TNF blockade may also affect B cell responses as originally suggested by the TNF-deficient mouse, which lacks primary B cell follicles and germinal centres upon immunisation with T cell-dependent (TD) antigens, resulting in impaired IgG antibody responses.4 More recently, we demonstrated that TNF selectively impaired the induction of TD alloantibodies, resulting in prolonged graft survival, without affecting splenic germinal centres.5 In humans, the antinuclear antibodies induced by infliximab are restricted to IgM reactivity towards double stranded DNA.6 The absence of IgG anti-dsDNA antibodies and reactivity against histones, nucleosomes or other nuclear proteins may point to a selective impairment of TD humoral responses.7 Additionally, RA patients treated by TNF blockade displayed reduced numbers of CD27 memory B cells and attenuated tonsillar germinal centre reactions.8 Therefore, we hypothesised that anti-TNF therapy blocks the induction of TD humoral responses by interfering with the germinal centre reaction. This study assessed this hypothesis by analysing the effect of TNF blockade on B cell activation, plasma cell differentiation, isotype switching, somatic hypermutation, and antibody responses to vaccination. We performed these experiments in SpA as, in contrast to RA, these patients do not display overt B alterations and have no concomitant treatment with methotrexate, azathioprine or corticosteroids, which could influence the B cell responses.9 ,10
Materials and methods
Patients and samples
Peripheral blood mononuclear cells (PBMCs) were obtained from 56 SpA patients fulfilling the European Spondyloarthropathy Study Group classification criteria.10 This cohort consisted of: (1) 20 patients treated with infliximab 5 mg/kg intravenously at week 0, 2 and 6 and thereafter every 8 weeks; (2) 10 patients treated with etanercept 50 mg subcutaneously every week; (3) 11 patients treated with adalimumab 40 mg subcutaneously every other week; and (4) 15 patients without TNF blockade. None of the patients received concomitant immunosuppressive treatment with methotrexate, azathioprine or corticosteroids. The demographic and clinical features of the patients are depicted in table 1. All patients gave written informed consent to participate in the study as approved by the Medical Ethics Committees of the Academic Medical Centre, Amsterdam and the Ghent University Hospital.
Demographic/clinical data of the spondyloarthritis patients
Flow cytometry
B and T cells were phenotyped by flow cytometry (FACScalibur, BD, San Jose, California, USA) using: FITC anti-IgD or CD45RA; PerCP-Cy5.5 anti-CD19 or CD4; APC anti-IgM, CD27 or CD3; PE-Cy7 anti-CD5; PE anti-IgG, HLA-DR, CD38, CD40, CD27 or CD28 BD, by BD Pharmingen, San Diego, California, USA); PE anti-CD45RO; PE-Cy7 anti-CCR7; AlexaFluor647 anti-CXCR5; AlexaFluor700 anti-CD3 and APC-eFluor780 anti-CD27 (eBiooscience, San Diego, California, USA). Analyses were performed on live CD19+ or CD3+CD4+ lymphocytes. IgD+CD27− B cells were defined as naïve, and CD27 B cells as memory. CD27 B cells were further divided in IgD+/−IgM+ or unswitched memory and IgG+ or switched memory. Results were expressed as percentage of positive cells or as mean fluorescence intensity.
Stimulation experiments
PBMCs were stimulated with (a) 100 ng/ml Phorbol 12-Myristate 13-Acetate (PMA) and 1 μg/ml Ionomycin (Sigma-Aldrich, Saint Louis, Michigan, USA) for 6 h as T cell stimulus; (b) 3 μg/ml CpG oligonucleotides ODNM362 (Invivogen, San Diego, California, USA) overnight as T cell-independent (TI) stimulus for B cells; or (c) 5 μg/ml anti-IgM (MH15 clone), 100 ng/ml CD40L (Enzo LifeSciences, Plymouth, Pennsylvania, USA) and 10 ng/ml IL-4 (R&D systems, Minneapolis, Minnesota, USA) for 3 days as TD stimulus for B cells. In some experiments, PBMCs were stimulated with 1, 3 and 10 μg/ml of recombinant TNF (R&D systems) for 6, 8 and 24 h. After incubation, the expression of costimulatory molecules was analysed by flow cytometry using FITC anti-IgD or CD45RA; PerCP-Cy5.5 anti-CD19 or CD4, APC anti-CD27 or CD3, PE anti-CD70, CD80, CD86, OX40L, CD154, OX40 or ICOS (BD Pharmingen).
In vitro differentiation to plasma cells
B cells were purified from PBMCs using anti-CD19 antibody conjugated magnetic beads (Miltenyi Biotec GmbH, Bergisch-Gladbach, Germany) and cultured with 100 ng/nl CD40L (Enzo LifeSciences), 50 U/ml IL-2 (Peprotech, RockyHill, New Jersey, USA) and 50 ng/ml IL-21 (R&D systems) for 2 days. After 48 h, the medium was replaced by fresh Rosewell Park Memorial Institute (RPMI) culture medium containing 50 U/ml IL-2 and 50 ng/ml IL-21 for another 48 h. Plasmablasts were quantified as CD20lowCD38high cells by flow cytometry using FITC anti-IgD, PE anti-CD38, PerCP-Cy5.5 anti-CD20 (BD Pharmingen) and APC-eFluor 780 anti-CD27 (eBiosciences).
IgκRHEMA assay
Somatic hypermutation was determined with the IgκRHEMA assay.11 Briefly, mRNA was extracted from PBMCs frozen in Trizol (Invitrogen, Carlsbad, California, USA) and cDNA synthesis was performed with the Revert-AidMinus FirstStrand cDNA synthesis kit (Fermentas, Burlington, Canada). The CDR1 locus in the variable region of the κ-light chain was amplified by PCR and digested with Fnu4HI and DdeI restriction enzymes. The length of the resulting fragments was determined using an ABIPrism-3100 Genetic Analyser (Applied Biosystems/Life Technologies, Carlsbad, California, USA). A 106-109 bp length indicated cleavage in CDR1 region, whereas 244 bp indicated no cleavage due to somatic hypermutation. The proportion between the 244-bp fragment peak area and the sum of the peak areas of 244-bp, 106-bp and 109-bp fragments indicated the degree of somatic hypermutation.
Vaccination
Ten patients treated with infliximab, 10 treated with etanercept and 10 patients without TNF blocker were vaccinated with Engerix-B (GlaxoSmithKline, Brentford, UK), a TD vaccine to Hepatitis B, and Pneumovax-23 (Merck, Whitehouse Station, New Jersey, USA), a TI vaccine to S. pneumoniae. Vaccination occurred 12 weeks after start of therapy. For Engerix-B, the initial vaccination was followed by boost vaccinations at week 6 and week 22. IgG antibodies to Pneumovax were determined with ELIZEN Pneumococcus kits (ZenTech, Liege, Belgium) following the recommendations from the manufacturer. Antibodies to Engerix were determined using ETI-AB-AUK Plus kits (DiaSorin, Saluggia, Italy).
Statistics
Values were presented as medians (IQR) and compared by non-parametric Wilcoxon's signed rank tests. p values <0.05 were considered statistically significant.
Results
TNF Blockade does not impair the activation and expression of costimulatory molecules by B cells
As TNF is an autocrine growth factor for human B lymphocytes,12 we first assessed whether TNF directly activates B cells. Unstimulated and stimulated (anti-IgM antibodies, CD40L and IL-4 for 20 h) PBMCs from healthy donors were cultured with medium or TNF (10 μg/ml). The percentage of cells positive for the activation marker CD69 was not affected by TNF (figure 1A). Stimulation with anti-IgM antibodies, CD40L and IL-4 strongly activated B cells as reflected by increased CD69 expression, but this activation also occurred in the presence of TNF (figure 1B). Similarly, B cells upregulated CD80 after stimulation but this upregulation was independent of TNF (figure 1C,D).13 ,14

(A) Expression of the early activation antigen CD69 by B cells cultured in medium alone (line histogram) or supplemented with TNFa (solid histogram), or (B) stimulated with anti-IgM, CD40L and IL-4 alone (line histogram) or supplemented with TNFa (solid histogram) (n=2). C) Expression of the costimulatory molecule CD80 by B cells cultured in medium alone (line histogram) or supplemented with TNFa (solid histogram), or (D) stimulated with anti-IgM, CD40L and IL-4 alone (line histogram) or supplemented with TNFa (solid histogram) (n=2).
Shorter experiments with lower doses of TNF showed marginal induction of CD69 and CD80 expression after 6 h (see onlinesupplementary figure S1), which subsided at later time points. Assessment of other B cell activation and costimulation markers (HLA-DR, CD40, CD86 and OX40L) after 24 h of TNF stimulation revealed a very small increase in the expression of CD86 (see onlinesupplementary figure S2). Taken together, these observations indicate that TNF stimulation does not promote marked B cell activation.
To compare the effects of TNF stimulation in vitro with the effects of TNF deprivation, we next analysed the ex vivo expression of activation markers in B lymphocytes from anti-TNF treated patients (infliximab, n=8) and controls (n=8). HLA-DR expression increased from 162 (149–290) at baseline to 211 (172–355) at week 12 (p=0.04). CD40 expression increased from 8.2 (7.3–15.3) at baseline to 13.7 (8.1–17.5) at week 12 (p=0.05). Experiments with samples from patients and controls (infliximab, n=7; controls=5), demonstrated that the expression of CD86 (54% (10–71%) vs 77% (65–83); p=0.001) and OX40L (15% (12–20) vs 19% (16–20); p=0.030) increased on CpG stimulated B cells obtained from baseline versus week 12 of TNF blockade (figure 2A-D, see onlinesupplementary table S1). These changes were specific to TNF blockade as no modulation was observed in the controls (see onlinesupplementary table S1 andfigure S3). The expression of costimulatory molecules was, however, not affected by TNF blockade in similar experiments with anti-IgM, CD40L and IL-4 stimulation to mimic TD B cell activation (data not shown). In summary, these experiments demonstrate that TNF blockade does not impair the capacity of B lymphocytes to get activated and upregulate costimulatory molecules, although the expression level may vary according to the type of activation.

Expression of HLA-DR (A) and CD40 (B) by CD19 B cells in peripheral blood mononuclear cells (PBMCs) collected at baseline and week 12 of TNF blockade in spondyloarthritis (SpA) (n=8). Percentage of CD86 (C) and OX40L+ (D) CD19 B cells at baseline and 12 weeks of TNF blockade in SpA (n=7). Data are represented as median and IQR.
TNF Blockade does not impair plasma cell differentiation
As TNF activates phosphorylation of Stat3 and Stat5b in B cells, thus controlling plasma cell differentiation,15–17 we next assessed whether TNF blockade modulated the differentiation of B cells to plasma cells. In vitro culture with CD40L, IL-2 and IL-21 induced a CD20lowCD38high plasma cell phenotype in 33% (22–39) and 23% (12–33) of the B cells obtained at baseline and week 12 of anti-TNF treatment (adalimumab, n=11), respectively (figure 3A). These results indicate that TNF blockade does not affect the differentiation towards plasmablasts.

(A) Plasmablast (CD20lowCD38highCD19) differentiation in vitro upon IL-2, CD40L and IL-21 stimulation of purified B cells collected from spondyloarthritis (SpA) patients at baseline and after 12 weeks of adalimumab treatment (n=11). The percentages of (B) IgD+CD27− naive B cells, (C) IgD+/− CD27 memory B cells, (D) unswitched IgM+CD27 memory B cells, and (E) CXCR5+ B cells were determined in peripheral blood mononuclear cells (PBMCs) obtained at baseline or after 12 weeks of TNF blockade in SpA patients (infliximab n=11, adalimumab n=11). Data are represented as median and IQR. NS: not significant.
TNF Blockade increases the percentage of circulating unswitched memory B cells and impairs somatic hypermutation
In germinal centre responses, recently activated IgD+CD27− B cells migrate towards the lymphoid follicles to interact with T cells and mature towards class-switched IgD-CD27 memory B cells. Alternatively, activated B cells can mature to unswitched IgM+CD27 memory B cells in extrafollicular sites or incomplete germinal centre reactions.18–20 To assess whether TNF blockade affects class switching, we phenotyped circulating B cells in anti-TNF treated patients (infliximab, n=11; adalimumab, n=11) and in control individuals (n=11). The frequency of naïve IgD+CD27− B cells decreased from baseline (53% (44–64) to 46% (37–57) after 12 weeks of TNF blockade (p=0.02) (figure 3B), whereas the memory IgD+/−CD27 subset, comprising both unswitched and switched cells, increased significantly from 33% (24–40) at baseline to 43% (28–52) at week 12 (p=0.004) (figure 3C). The frequency of these subsets remained stable in the control group (data not shown). A more detailed analysis of switched and unswitched memory cells, revealed a significant increase of the IgD+/−IgM+CD27 unswitched memory B cells after TNF blockade (17% (15–21) at baseline vs 23% (17–28) at week 12; p=0.04)(adalimumab, n=11) (figure 3D). This effect was specific for TNF blockade, as the frequency of this B cell subset did not change over time in the control SpA patients (n=5) (data not shown). No changes were observed in the percentages of switched memory B cells, neither in treated nor control patients. The expression of CXCR5, crucial for migration towards the lymphoid organs,21 ,22 was not affected by anti-TNF treatment (adalimumab, n=11) (81% (57–89) positive cells at baseline vs 83% (73–90) at week 12) (figure 3E). As a normal germinal centre reaction also requires appropriate activation and influx of T cells, we additionally phenotyped the circulating T cells before and after anti-TNF (adalimumab, n=11). We did not observe changes in the circulating T cell subsets as defined by CD4, CD8, CD45RO and CD45RA or in the expression of CXCR5, CD40L, CD27, CD28, OX40 and ICOS (data not shown).
Somatic hypermutation is the second key feature of germinal centre-dependent B cell maturation. We therefore measured the level of somatic hypermutation in circulating B cells before and 38 weeks after anti-TNF treatment (infliximab, n=17).11 In the anti-TNF treated SpA patients, the frequency of mutated transcripts decreased from 82% (73–85) at baseline to 74% (68–79) at week 38 (p=0.04). In contrast, the level of somatic hypermutation remained stable in the SpA patients without TNF blockers (n=9). The increase in unswitched circulating memory B cells and the decrease of somatic hypermutation suggests that TNF blockade affects the germinal centre reaction.
TNF Blockade abrogates the induction of TD humoral responses
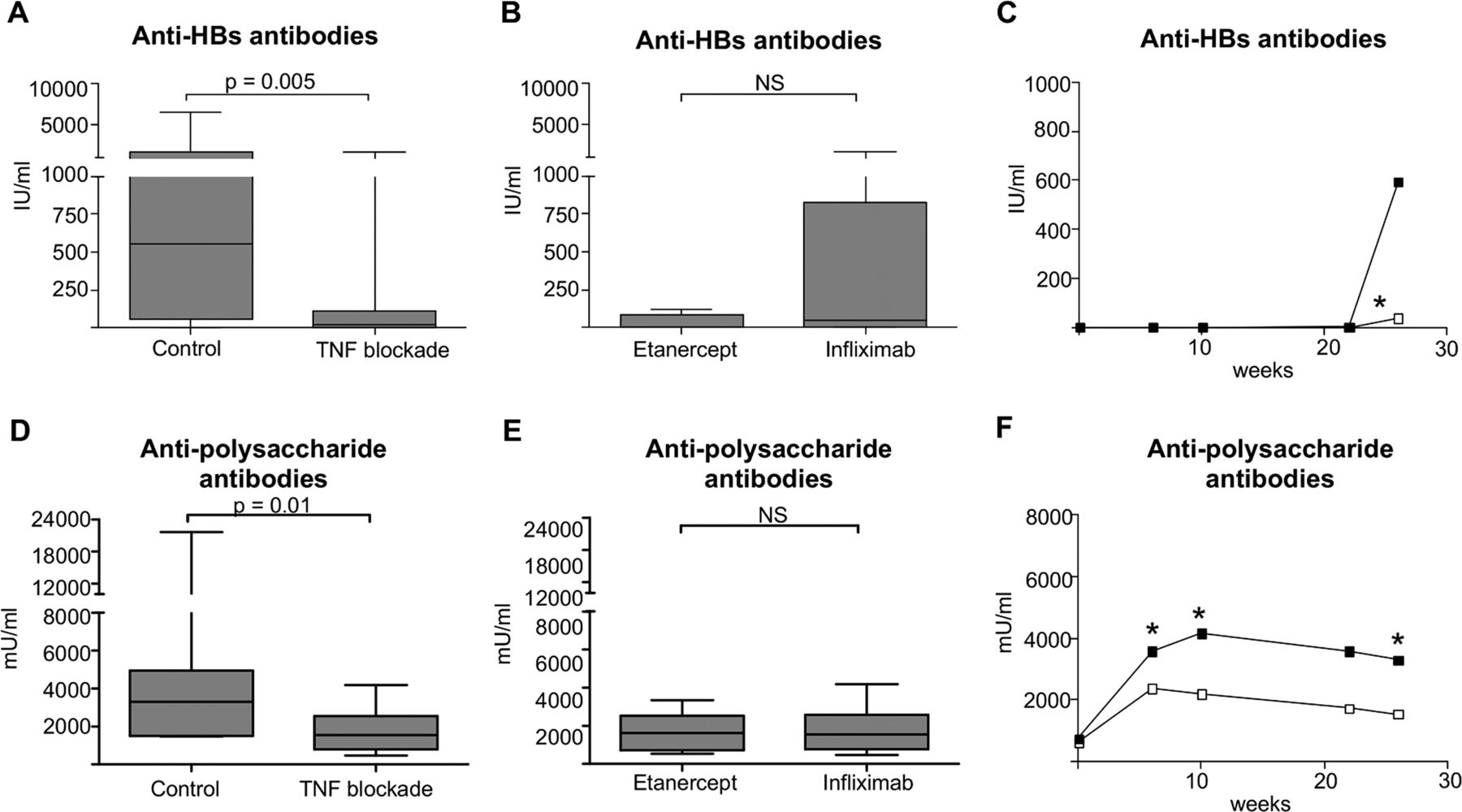
Although we had no access to lymphoid tissues to analyse the germinal centres in these patients, we assessed whether TNF blockade impairs their function by analysing the humoral response to primary TD vaccination against hepatitis B. SpA patients without TNF blockade (n=10) showed a clear humoral response at week 26, with median antibody titres of 595 IU/ml (65–2190 IU/ml). In contrast, the median antibody concentration was only 10 IU/ml (0–115 IU/ml) in the patients with TNF blockade (n=20) (p=0.005) (figure 4A,C). In fact, only four of 20 patients developed a robust response to the vaccination (titres>120 IU/ml) and had similar affinity to the response in controls, as measured by dilution ELISA (see onlinesupplementary figure S4). We did not observe differences in antibody responses between patients treated with infliximab (n=10) and etanercept (n=10) (figure 4B). To ascertain that the impaired response to hepatitis B was not due to overall immunosuppression, the same patients were simultaneously vaccinated with a TI pneumococcal polysaccharide vaccine. In line with previous reports,23 24 26–28 TI vaccination induced a clear antibody response in treated patients, although the IgG titres were significantly lower than in the controls (1545 mU/ml (794–2545 mU/ml) vs 3300 mU/ml (1498–4943 mU/ml) at week 26)(p=0.01) (figure 4D,F). Also for the pneumococcal polysaccharide vaccination, we did not observe differences between infliximab and etanercept treated patients (figure 4E). Taken together, these data indicate that TNF blockade severely impedes the humoral response to primary TD immunisation in arthritis patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Control (n=10) or anti-TNF treated spondyloarthritis (SpA) patients (n=10 with infliximab or etanercept) were vaccinated with Engerix-B, and Pneumovax. Serum was collected at baseline and 6, 10, 22 and 26 weeks after vaccination. Vaccine antibody titres were determined by ELISA. HBs antibody levels of control and treated patients at week 26 (A), infliximab and etanercept patients at week 26 (B); HBs antibody titres depicted in time (C). Polysaccharide antibody levels of control and TNF treated patients at week 26 (D), and infliximab and etanercept patients at week 26 (E); Polysaccharide antibody titres depicted in time (F). Data is represented as median+IQR. NS: not significant.
Discussion
There is circumstantial evidence indicating that TNF blockade may directly modulate humoral responses.5 ,8 This study provides direct evidence for this hypothesis by demonstrating a severe defect in the humoral response to primary vaccination against hepatitis B in anti-TNF treated SpA patients. Our study was conducted in SpA rather than RA patients to avoid biases related to altered B cell biology29 and use of concomitant immunomodulatory drugs,25 ,27 but the results are in agreement with two abstract reports in RA. The first study reported a 100% response rate to hepatitis B vaccination in methotrexate treated RA versus a 29% response rate in etanercept treated RA.30 The second study reported that only one of 25 RA and three of 27 SpA patients on infliximab treatment displayed a good response to hepatitis B vaccination.31 Taken together, these data demonstrate that TNF blockade profoundly affects the response to hepatitis B vaccination independently of disease background and type of TNF blocker.
As the induction of TD humoral responses is dependent on germinal centre reactions, our findings are also in agreement with studies on the effect of TNF blockade on germinal centre reactions. First, TNF-knockout mice lack germinal centres and develop impaired IgG responses upon immunisation with TD antigens.4 Second, ectopic lymphoid structures in inflamed synovium disappear upon TNF blockade.32 ,33 Furthermore, a histological analysis of tonsils indicated a significant decrease of the number and size of germinal centres in four RA patients treated with etanercept in comparison with five healthy controls..8 In our study, TNF blockade altered two key functional features of germinal centres: isotype switching of memory B cells and somatic hypermutation, strongly supporting the hypothesis of TD responses being impaired at the level of the germinal centre reaction. Anolik et al8 suggested that the impact of TNF blockade on germinal centres was related to a decrease in follicular dendritic cells. A study with lymphotoxin blockade in primates, however, demonstrated that interference with follicular dendritic cell networks does not necessarily abrogate germinal centre formation.34 Accordingly, our recent study on TNF blockade in rat allografts5 demonstrated impaired TD responses without any significant effect on the number or size of splenic germinal centres. In the present study, we could not investigate the histology of germinal centres, as we did not have access to lymphoid tissues of our SpA patients. However, our in vitro and ex vivo analyses of circulating lymphocytes demonstrated no intrinsic defect of B cells in anti-TNF treated patients as there was no impairment in B cell activation, costimulatory molecule expression and plasmablast differentiation. Similarly, we did not observe significant alterations in T cell populations. These data support a selective functional impairment of the germinal centre reaction during TNF blockade rather than a general B or T cell defect. Deciphering the exact mechanisms would require mouse studies in which antigen-specific B cells and their interactions with follicular helper T cells and dendritic cells can be visualised during humoral responses.
These considerations raise the question whether TNF blockade only impairs the induction of primary TD responses or also affects TI primary response and recall responses. Multiple studies investigated extensively the TI response to pneumococcal vaccination during TNF blockade. Although not completely consistent because of differences in disease background, co-medication and read out, most studies indicate no25 ,26 ,35 or moderate23 ,27 ,28 ,36 impairment of the vaccination response. In the present study, we observed a moderate decrease in antibody titres to pneumococcal polysaccharides, albeit much less pronounced than for the TD vaccination against hepatitis B in the same patients. As to TD responses, most clinical studies were performed with influenza vaccination25 26 37–39 and consistently reported that the response was impaired by TNF blockade. Moreover, these reports39 suggest that this defect is mainly seen in primary responses.
The differential effect of TNF blockade on TD and TI humoral responses questions the clinical relevance of our findings. In clinical vaccination studies, the small effect on pneumococcal vaccination and moderate effect on (partially recall) influenza vaccination does not prevent that most patients reach protective antibody titres. In the case of a primary TD vaccination as performed in this study, however, the impairment is profound and of obvious clinical relevance. Hence our study strongly pleads for the administration of primary TD vaccinations before the initiation of TNF blockade. Alternatively, adapted vaccination schedules (dosage, adjuvant, number of boost vaccinations) could overcome the anti-TNF induced impairment. The clinical relevance, however, is not restricted to protective responses to infectious agents. As illustrated by our rat allograft study, this novel mechanism of action of TNF blockers may also be beneficial in specific situations such as the development of alloantibodies after transplantation5 or epitope spreading in different types of autoimmune disease. Finally, it is tempting to speculate that this mechanism may contribute to protect patients from developing a humoral response to the anti-TNF drugs themselves as despite repetitive administration of these non-self proteins most patients do not develop antidrug antibodies.
In conclusion, we demonstrate in human arthritis patients that TNF blockade severely impedes the induction of primary TD humoral responses, most probably by interference with germinal centre responses. This finding warrants further investigation of the exact cellular and molecular basis of this novel mechanism of action of TNF blockade.
Acknowledgments
This study was partially supported by the Dutch Arthritis Foundation (Reumafonds). Leen De Rycke was supported by a VENI grant of The Netherlands Organization for Scientific Research (NWO). Dominique Baeten was supported by a VIDI grant of The Netherlands Organization for Scientific Research (NWO). The authors thank Dr Filip Van den Bosch and Professor Dr Filip De Keyser for referring patients.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Contributors 1A Study design: DB, GFS, LDR, PPT. 1B/Acquisition of data: GFS, BB, JP. 1C/Analysis and interpretation of data: DB, GFS, MB, TC. 2A/Drafting the article: GFS. 2B/Revising the article: GFS, TC, DB, LDR, PPT, BB, MB, JP. 3A/Approval of the final article: GFS, TC, DB, LDR, PPT, BB, MB, JP.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.