





Abstract
This study examined inflammatory responses from primary cultured human bronchial epithelial cells in chronic obstructive pulmonary disease (COPD) and the clinical factors modulating them.
Epithelial cells from bronchoscopic biopsies from 14 patients with COPD ((mean±sd) age 74.6±5.7 yrs, forced expiratory volume in one second (FEV1) 1.21±0.36 L, FEV1 % predicted 51.1±15.8%, 51.5±24.0 pack-yrs of smoking, inhaled steroid dosage 1237.5±671.0 µg·day−1, Medical Research Council (MRC) dyspnoea score 3.18±1.33) and eight current/exsmokers with normal pulmonary function (age 60.4±13.5 yrs, FEV1 2.66±1.27 L, FEV1 % pred 89.6±17.7%, 49±44 pack-yrs of smoking, MRC dyspnoea score 1±0) were grown in primary culture and exposed to 50 ng·mL−1 tumour necrosis factor‐α.
Stimulated COPD cells produced significantly more interleukin (IL)‐6 at 24 and 48 h, and IL‐8 at 6 and 24 h than unstimulated COPD cells. This response was not seen in cells from current/exsmokers. IL‐6 and IL‐8 production was lower in COPD patients taking inhaled steroids.
Following an inflammatory stimulus, bronchial epithelial cells in chronic obstructive pulmonary disease show a significant cytokine response not seen in smokers with normal pulmonary function and this may be modified by inhaled steroid therapy.
This study was supported by the Joint Research Board, St Bartholomew's Hospital, London, UK.
Patients with chronic obstructive pulmonary disease (COPD) exhibit heightened airway inflammation, characterised by increased levels of interleukin (IL)‐8 and tumour necrosis factor (TNF)‐α in induced sputum 1, a predominantly neutrophilic cellular infiltrate in the airway lumen 2, and increased lymphocyte numbers in the submucosa and alveolar parenchyma 3. They are prone to exacerbations, which are a major cause of hospital admission 4 and an important determinant of health-related quality of life 5. It has been demonstrated that stable patients with a history of frequent exacerbations have increased induced sputum levels of IL‐6 and IL‐8, and that sputum levels of IL‐6 further increase at COPD exacerbation 6. This suggests that COPD may be associated with enhanced airway epithelial cell activation, particularly at exacerbation, though the mechanisms underlying this are largely unknown.
There is increasing evidence that as well as acting as an important physicochemical barrier protecting the submucosa, the airway epithelium is a metabolically active mediator in the pathophysiology of respiratory disease 7. Bronchial epithelial cells synthesise and release a number of pro-inflammatory factors both constitutively and in response to external stimuli 9, thereby influencing inflammatory cell chemotaxis, recruitment, activation and differentiation 10. However, the role of the bronchial epithelium in modulating inflammatory processes in COPD, either under stable conditions or at exacerbation, has not been identified.
This study was designed to evaluate the response made by primary-cultured human bronchial epithelial cells (HBEC) from patients with COPD to an inflammatory stimulus. TNF‐α is a pro-inflammatory cytokine present in the COPD airway 11, which stimulates cytokine release by epithelial cells 12. Stimulation with TNF‐α was therefore used as a model for what may occur, in part, at the epithelial level during COPD exacerbations. In addition, factors modulating epithelial inflammatory responses in COPD were examined, including physiological parameters and inhaled steroid therapy.
Materials and methods
Study subjects
Volunteers (n=22) were prospectively recruited to the study from those patients undergoing routine clinically indicated bronchoscopy at the London Chest Hospital, UK. Fourteen patients with COPD and eight current or exsmokers with normal pulmonary function were sampled. COPD was defined as a forced expiratory volume in one second (FEV1) <70% predicted for age and height, β2‐agonist reversibility on predicted FEV1 of <15% and/or 200 mL with airflow obstruction as evidenced by an FEV1/forced vital capacity (FVC) ratio of <70%. Patients were excluded from the study if they had active respiratory tract infections, including ongoing or recent exacerbations of COPD, suspected tuberculosis, bronchiectasis or asthma. Before the procedure, a detailed history was taken from each study subject outlining their smoking habits (current smoking status, years of smoking and number of packs of 20 cigarettes smoked per day), drug history including inhaled steroid dosage, daily stable respiratory symptoms and Medical Research Council dyspnoea score. Baseline spirometry, and, where possible, arterialised capillary earlobe blood gases 13 were also obtained prior to the planned procedure.
Table 1⇓ shows the physiological characteristics of the study subjects. The COPD patients were older than the normal subjects (p<0.05). The two groups were matched for pack-yrs of smoking (p=0.15), however, eight (51.7%) of the COPD patients and six (75%) of the normal subjects were current smokers. Within the COPD group, eight (51.7%) patients were taking regular inhaled steroids at a mean±sd dose of 1,237.5±671 µg·day−1. Within the current/exsmokers with normal pulmonary function, two (25%) patients had a final diagnosis of bronchial carcinoma and one (12.5%) was found to have a mesothelioma. None of the patients with COPD had bronchial carcinoma and none of the subjects were taking any medications (other than inhaled steroids) that may have affected their airway inflammatory responses.
Physiological characteristics of study subjects
Each volunteer underwent fibreoptic bronchoscopy under light sedation and local anaesthesia according to the British Thoracic Society guidelines 14. Endobronchial biopsies were taken using rotatable cup-biopsy forceps (Olympus FB-19KR; KeyMed Ltd, Southend-on-Sea, UK) at the level of a segmental or subsegmental carina on the side opposite to any radiological abnormality precipitating the bronchoscopy. Biopsies were immediately placed in ice-cold medium 199 (Sigma-Aldrich Co Ltd, Poole, UK) containing 1% gentamicin (Roussel Laboratories Ltd, Uxbridge, UK) and processed for tissue culture within 30 min.
The Ethics Committee of the East London and City Health Authority granted full approval for this study and fully informed written consent was obtained from all participants prior to their inclusion.
Materials
All reagents and chemicals were of tissue culture grade and provided by the Sigma-Aldrich Co Ltd unless otherwise stated. All prepared solutions were filtered through a 0.20 µm pore Minisart syringe filter (Sartorius Ltd, Epsom, UK) before addition to the cell cultures.
Culture of bronchial epithelial cells
HBEC were grown in primary culture using the explant cell culture technique developed in the laboratory. The epithelium was dissected away from any underlying lamina propria, cut into smaller sections ∼0.5 mm3 in size and washed three times with sterile medium 199. Single sections of epithelium were then explanted onto 6‐cm Falcon Primeria plastic culture dishes (Becton Dickinson Ltd, Oxford, UK) and incubated with 1.5 mL of complete culture medium at 37°C in a humidified 5% carbon dioxide air atmosphere. The culture medium was prepared by mixing 250 µg bovine pancreatic insulin, 250 µg of human transferrin, 1.5 mL of antibiotic/antimycotic solution, 3 mg l‐glutamine and 2.5 mL NU-serum (Universal Biologicals Ltd, Gloucestershire, UK) in 100 mL of medium 199. The medium in each dish was replaced with maintenance medium after 7 days and every 48 h thereafter.
Exposure of human bronchial epithelial cells to TNF‐α
The cultures were observed until the cells had grown to confluence after 3–4 weeks. On the day prior to the experiment, explants were removed and cultures were washed, and incubated overnight in medium 199 and an antibiotic/antimycotic solution for 24 h. HBEC cultures were then washed twice with 2 mL of sterile medium 199 before the addition of the appropriate reagents. In the case of TNF‐α‐exposed dishes, this consisted of 2.5 mL of a 50 ng·mL−1 solution of recombinant TNF‐α (R&D Systems Europe, Abingdon, UK) made up in medium 199 and antibiotic/antimycotic solution. This concentration of TNF‐α had been determined not from physiological measurements, but using preliminary exposure experiments in the laboratory showing inadequate stimulation at 25 ng·mL−1 but cell death with reduced cell product at 100 ng·mL−1 15. The unstimulated dishes were incubated with 2.5 mL of medium 199 and antibiotic/antimycotic solution only. An aliquot of supernatant was collected from culture dishes immediately after the addition of either TNF‐α or control solution (time zero). Cells were then incubated as before, and aliquots of supernatant were collected at 6, 24 and 48 h. All samples were stored at −70°C for cytokine analysis using quantitative sandwich immunoassay techniques (R&D Systems Europe). IL‐6, IL‐8 and soluble intercellular adhesion molecule (sICAM)‐1 levels were measured in the cell supernatant. Experiments were terminated at 48 h, at which time cells were stored at −70°C for total cellular protein analysis as described by Lowry et al. 16.
Analysis
Cytokine release from HBEC was measured in absolute values of pg·µg cellular protein−1 at each time point. To allow for differences in baseline cytokine production, the percentage change in cytokine production from time zero was calculated for times 0–6, 0–24 and 0–48 h for each experimental group. Normally distributed data were summarised by mean±sd and skewed data by medians (interquartile range (IQR)). Continuous variables with normal distributions were compared by an unpaired t‐test, whereas those with non-normal distributions were compared by the Mann-Whitney U‐test or Wilcoxon signed-rank test, as appropriate. Cytokine levels, and physiological and other indices were correlated using Spearman's rank correlation.
Results
Culture and identification of human bronchial epithelial cells
The identity of the epithelial cells was confirmed in all cultures by light microscopy, demonstrating large numbers of cells with polygonal morphology. In randomly selected cultures, electron microscopy and immunocytochemical staining for cytokeratin were performed using monoclonal antibody preparation CAM 5.2 (Becton Dickinson Ltd) to confirm the epithelial nature of the cells. Only cultures demonstrating ≥50% ciliated cells, with good ciliary motility, were used in experiments, and no qualitative differences in the degree of cilliation or differentiation were seen in this study between COPD cells and those from subjects with normal lung function. Staining with specific monoclonal antibodies against contaminating cell types, such as fibroblasts, did not show any evidence of these cells.
Cytokine production by human bronchial epithelial cells in the absence of TNF‐α stimulation
sICAM‐1 production by COPD HBEC under baseline conditions was lower in the presence of inhaled steroid therapy (rho=−0.732, p=0.039 for sICAM‐1 at 24 h). No relationships were seen in normal HBECs between baseline cytokine production and any of the physiological or clinical parameters outlined above.
Cytokine response to TNF‐α stimulation: COPD versus current/exsmoker cell cultures
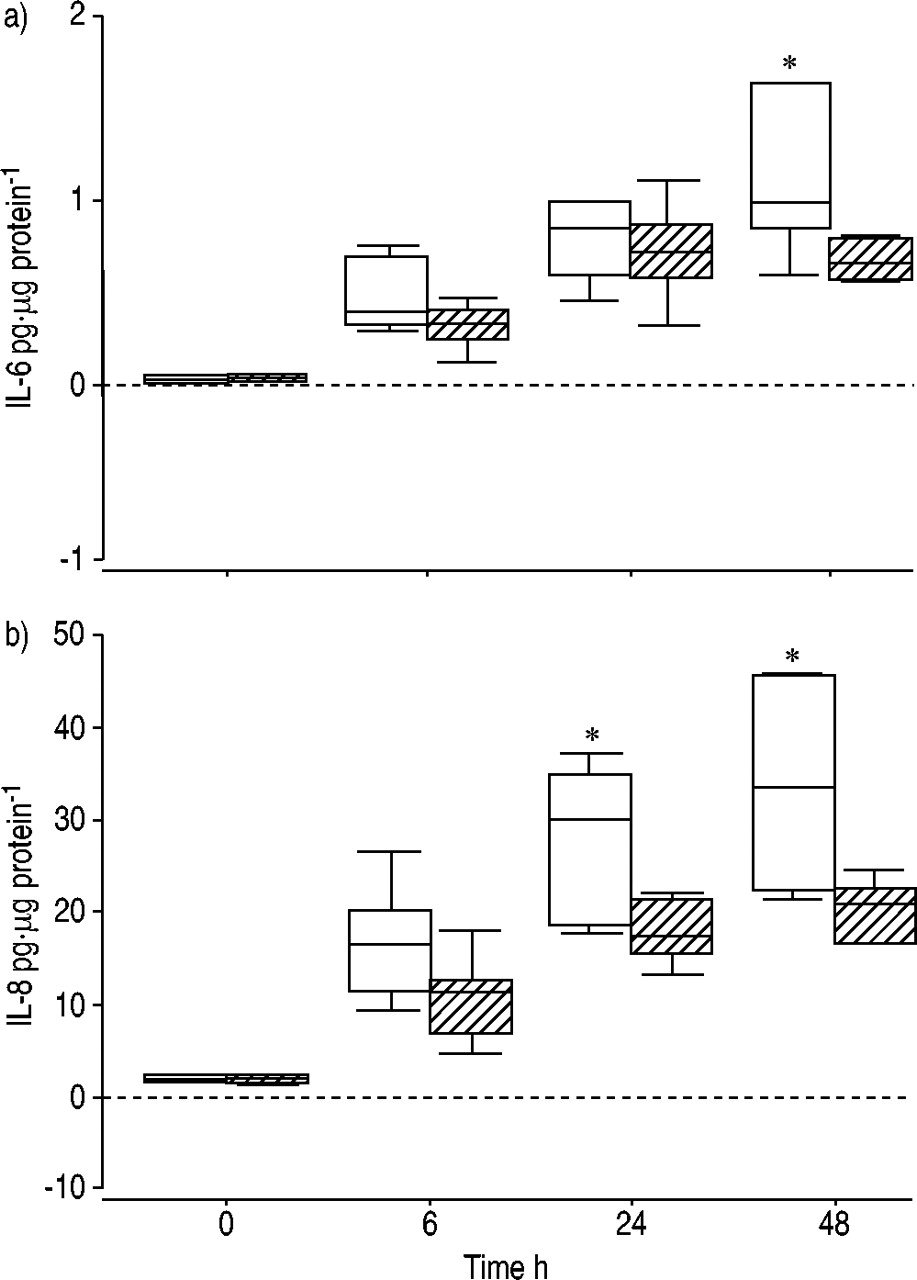
Table 2⇓ shows the median (IQR) levels of IL‐6, IL‐8 and sICAM‐1 production at each time point from unstimulated cells and cells exposed to TNF‐α within the smokers/exsmokers without airways obstruction, and cells from COPD patients. The absolute level of cytokine production was greater from cells from current/exsmokers than from COPD cells at each time point, irrespective of stimulation with TNF‐α. However, within the current/exsmokers there was no statistically significant response to TNF‐α, as measured by an increase in cytokine production by HBEC over baseline values. Within the COPD group, TNF‐α‐exposed HBEC produced significantly greater amounts of IL‐6 at 6 (p=0.053), 24 (p=0.006) and 48 h (p=0.015) compared with unstimulated cells (fig. 1a⇓). Similarly, IL‐8 production was greater at 6 (p=0.025) and 24 h (p=0.045) in the stimulated group in contrast to unstimulated cells (fig. 1b⇓). A trend towards increased sICAM‐1 production in the TNF‐α‐exposed COPD cells was seen at 48 h (p=0.08; fig. 1c⇓).

Production of a) interleukin (IL)‐6, b) IL‐8 and c) soluble intercellular adhesion molecule (sICAM)‐1 by control (□) and tumour necrosis factor (TNF)‐α‐stimulated (└), primary-cultured chronic obstructive pulmonary disease bronchial epithelial cells. The boxes represent the interquartile range containing 50% of values. The bars extend to the highest and lowest values, excluding outliers, and the horizontal lines indicate the median values. *: p<0.05 comparing TNF‐α‐stimulated and unstimulated cells.
Interleukin (IL)‐6, IL‐8 and soluble intercellular adhesion molecule (sICAM)‐1 production
When the percentage changes in cytokine production from time zero were calculated, a significant increase in sICAM‐1 production in response to TNF‐α was seen in the COPD group at 48 h (p=0.014).
Effect of inhaled steroid therapy on cytokine production from stimulated COPD human bronchial epithelial cells
Figure 2⇓ shows IL‐6 and IL‐8 production from stimulated COPD cell cultures in the presence and absence of regular, inhaled steroid therapy. Median (IQR) IL‐6 production at 48 h in the steroid-free group was 0.98 (1.35) in contrast to 0.72 (0.21) pg·µg cellular protein−1 in those on inhaled steroids (p=0.03). IL‐6 production was negatively correlated with inhaled steroid dosage at all time points (time 0–6 h: rho=−0.849, p=0.016; 0–24 h: rho=−0.926, p=0.003; 0–48 h: rho=−0.949, p=0.05). IL‐8 production at 24 h was 30.0 (17.1) in the steroid-free group and 19.50 (7.75) pg·µg protein−1 in the steroid-treated group (p=0.03). At 48 h, IL‐8 production was 33.6 (23.4) and 21.7 (6.3) pg·µg protein−1, respectively (p=0.05). No direct relationship was seen between IL‐8 levels and inhaled steroid dosage. Inhaled steroids did not affect sICAM‐1 production at any time point after stimulation.
{kind=link}
{kind=link}
Production of a) interleukin (IL)‐6 and b) IL‐8 by tumour necrosis factor‐α‐stimulated, primary-cultured chronic obstructive pulmonary disease bronchial epithelial cells in the presence (└) and absence (□) of regular, inhaled steroid therapy. The boxes represent the interquartile range containing 50% of values. The bars extend to the highest and lowest values, excluding outliers, and the horizontal lines indicate the median values. *: p≤0.05.
Inhaled steroid therapy was related to spirometric criteria; the mean±sd FEV1 in patients taking inhaled steroids was 0.98±0.08 in contrast to 1.50±0.41 L in those not taking them (p=0.027). Similarly the mean FEV1 % pred in the steroid-treated group was 40.0±9.4 in contrast to 66 (2.9) in the steroid-free group (p<0.001), and the mean FEV1/FVC ratio was 38 (9.5) and 61 (7.5) (p<0.001) in the two groups, respectively.
Discussion
This study examined the inflammatory responses made by primary-cultured bronchial epithelial cells in COPD, and the clinical and physiological factors modulating these in a well-characterised group of patients. Compared with current/exsmokers without airways obstruction, epithelial cells from patients with COPD were found to release lower levels of IL‐6, IL‐8 and sICAM‐1 under baseline conditions, but mounted a significant cytokine response to TNF‐α, which was not seen in cells from normal smokers. Within the COPD group, the IL‐6 and IL‐8 response was lower in cells from patients taking inhaled corticosteroids.
It is interesting that in this study, both under baseline conditions and after TNF‐α stimulation, lower absolute cytokine levels were detected from cells from COPD patients than from smokers with normal pulmonary function. This could be due to a defect in cytokine production by COPD HBEC in primary culture or a reflection of the situation in vivo. The explant cell culture technique developed in the laboratory is an established and validated method, which has previously been shown to allow the growth of well-differentiated epithelial cells with similar morphological and biochemical characteristics to those found in vivo 17. However, phenotypic differences between cells from COPD patients and those from normal subjects could have affected the results reported. Bronchial biopsies have shown epithelial shedding, reduced numbers of cilia 18 and a reduction in the proportion of ciliated cells 19 in chronic bronchitis and COPD. In a previous study of bronchial epithelial cells in culture, 10% of cells from smokers with normal lung function were reported to be ciliated, while those from patients with COPD were almost completely devoid of cilia 20. While this was not found in the present study, disparities in the state of differentiation of the cell cultures may have accounted, in part, for the differences in levels of cytokine production by HBEC from COPD cells and those from current/exsmokers. This would need to be examined in similar studies.
There is also evidence to suggest that constitutive cytokine production by airway epithelial cells may be downregulated in COPD. Schulz et al. 21 recently reported experimental results consistent with the present findings, with levels of constitutive IL‐8 production from COPD epithelial cells in primary culture, which were approximately one-third of that seen from normal cells. It was postulated that this may be due to altered post-translational processing 21. Factors may also exist in vivo that modulate airway inflammatory responses and that are absent in the sterile conditions of cell culture. The presence of colonising bacteria in the lower airway is increasingly recognised as an important independent stimulus to airway inflammation in COPD 22. The resultant recruitment of inflammatory cells 24, and interactions between bacterial and other cell products and the pulmonary epithelium 25, may account for the increased levels of inflammatory cytokines isolated in sputum and bronchoalveolar lavage fluid in COPD, and for the downregulated epithelial inflammatory responses seen in this study under baseline conditions. Most studies of airway inflammation in COPD have been performed using sputum or bronchial biopsies and little information is available regarding whether airway epithelial activity in this condition reflects inflammation seen in other compartments. This requires further evaluation.
In the presence of an inflammatory stimulus like TNF‐α, a significant increase in cytokine production over constitutive release was seen in COPD epithelial cells, but not in those from current/exsmokers with normal pulmonary function. A recent study, comparing the effects of cigarette smoke, demonstrated a similar lack of cytokine production in primary-cultured epithelial cells from normal smokers, while cells from patients with COPD once again showed a significant response 20. However, epithelial cells from normal subjects with normal lung function have previously been shown to produce good inflammatory cytokine responses to TNF‐α 27. These findings suggest that there may be important differences between the airway epithelium of smokers who do not develop COPD and those who do, with the former failing to mount a response to a common inflammatory stimulus. This could represent an important protective mechanism in smokers who do not develop COPD, possibly explained by differential expression of epithelial TNF‐α receptors. These were not examined in the present study, and further studies of primary bronchial epithelial cells in well-characterised normal smokers and patients with COPD are now required.
The enhanced cytokine production by COPD cells following TNF‐α stimulation may also be indicative of heightened airway inflammatory responses in this patient group. While not providing a complete model of COPD exacerbation, the response of the epithelium to a given inflammatory stimulus may be informative about general epithelial responses at COPD exacerbation. TNF‐α is a ubiquitous cytokine that forms part of the final common pathway of a number of inflammatory processes in the airway. Upregulation of IL‐6 and IL‐8 production in response to this inflammatory stimulus could affect key inflammatory processes in the pathophysiology of COPD, including enhanced chemotaxis of inflammatory cells into the airway and upregulation of mucin genes. This response may therefore be a component of the mechanisms governing COPD exacerbation and could partly explain the tendency to exacerbation in this patient group.
It is interesting that in this study no significant sICAM‐1 response was seen following TNF‐α stimulation, although previously TNF‐α was shown to enhance epithelial expression of this molecule 28, which is upregulated in bronchial biopsies from patients with COPD 29. These data therefore suggest that sICAM‐1 production by epithelial cells in COPD may be dependent on a number of cofactors. ICAM‐1 is the major receptor for human rhinovirus attachment on epithelial cells 30, and human rhinovirus has been shown to induce further ICAM‐1 expression, thereby promoting inflammatory cell recruitment and activation as seen in exacerbations 31. Airway epithelial cells play an important role in virus-induced inflammation 32 with viral infections, most commonly human rhinovirus, accounting for a significant proportion of COPD exacerbations 33. A limitation of this cell culture model is that it did not take into account the crucial role of common viral triggers at COPD exacerbation and the epithelial response to them. An examination of the cellular pathways behind this finding may increase the understanding of the mechanisms of exacerbation in COPD.
A potentially important finding in this study was the attenuation of the IL‐6 and IL‐8 response to TNF‐α seen in HBEC from COPD patients taking regular, inhaled steroid therapy. While in vitro evidence has demonstrated inhibitory effects of corticosteroids on IL‐6 and IL‐8 production in epithelial cell lines 35, as well as on neutrophil chemotaxis 37 and airway neutrophilia 38, clinical studies of inhaled steroid therapy in COPD have largely shown this class of drugs to be of low efficacy. However, it was shown recently that inhaled corticosteroids may reduce the severity of COPD exacerbations 39. On the basis of the current findings, the mechanism for this could be a reduction in exacerbation-associated inflammation. It has also been shown that inhaled steroids can reduce exacerbation frequency in COPD 40. In this study, lower constitutive release of sICAM‐1 was seen in cells from patients taking inhaled steroids. Therefore, it may be that inhaled steroids have a particular role in modifying virus-associated exacerbations, thereby reducing exacerbation frequency. Studies of steroid withdrawal in COPD have found a lag of several weeks between discontinuation of steroids and the next exacerbation 41, suggesting that the effects of corticosteroid therapy on mediator release, as seen in this study, are likely to have been the result of transcriptional alterations in cytokine production that persisted in vitro.
In summary, this study has demonstrated significant differences between primary airway epithelial cytokine production in patients with chronic obstructive pulmonary disease and smokers with normal pulmonary function, both constitutively and in response to an inflammatory stimulus. These observations may be important in distinguishing smokers who do not develop chronic obstructive pulmonary disease from those who do, as well as explaining the predisposition to exacerbation in this condition. The authors have also shown that epithelial inflammatory responses in chronic obstructive pulmonary disease can be modified by inhaled steroid therapy, which may partly explain the clinically observed effects of inhaled corticosteroids on chronic obstructive pulmonary disease exacerbations.
Acknowledgments
The authors would like to thank G. Berlyne and M. Beckles for assistance with obtaining the biopsy specimens.
- Received October 14, 2002.
- Accepted March 11, 2003.
- © ERS Journals Ltd
References









