Article Text

Abstract
Introduction Despite strong evidence that maturation patterns of the gut microbiome in early life influence the risk for childhood asthma, very little is known about gut microbiota patterns in adults with established asthma, and of greater interest relationships to phenotypic features that characterise asthma heterogeneity.
Methods Fifty-eight faecal samples from 32 adults with (n=24) and without (n=8) asthma were analysed using 16S ribosomal RNA gene sequencing methods to characterise intestinal bacterial composition. Compositional stability of paired samples was evaluated and features of gut bacterial community structure analysed in relation to extensive clinical characterisation data collected from subjects, who were enrolled in a prospective observational cohort study at the University of Michigan.
Results Differences in gut bacterial community structure were associated with aeroallergen sensitisation and lung function as assessed by forced expiratory volume in 1 s (FEV1) %predicted. Associations with FEV1 were consistently observed across independent analytic approaches. k-means clustering of the gut microbiota data in subjects with asthma revealed three different clusters, distinguished most strongly by FEV1 (p<0.05) and trends in differences in other clinical and inflammatory features.
Conclusion In this pilot study of asthmatic and non-asthmatic subjects, significant relationships between gut microbiota composition, aeroallergen sensitisation and lung function were observed. These preliminary findings merit further study in larger cohorts to explore possible mechanistic links to asthma phenotype.
- gut microbiome
- adult asthma
- phenotype
- lung function
- 16S rRNA
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
The role of the gut microbiome in adult phenotypes of asthma is underexplored and may contribute to disease heterogeneity.
In this study significant relationships were observed between differences in intestinal bacterial composition and characteristics of asthma, including lung function (forced expiratory volume in 1 s %predicted) and degree of aeroallergen sensitisation.
Results of this pilot study suggest gut microbiota associations with asthmatic phenotype in adults with established disease, supporting further investigation of mechanistic links in larger studies.
Introduction
Despite strong evidence that maturation patterns of the gut microbiome in early life influence susceptibility to childhood asthma and other allergic diseases,1 little is known about gut microbiota patterns in adults with established asthma,2 and of greater interest relationships to clinical features of asthma which is heterogeneous in adults. We and others have previously reported differences in the airway microbiome associated with phenotypic features of asthma, including bronchial hyper-reactivity, asthma control and airway inflammation patterns.3–7 Described phenotypes of asthma also include ones linked to markers of systemic inflammation8 and extrapulmonary comorbidities such as obesity,9 but mechanisms by which these affect lung disease are not fully known. Whether gut microbiome differences among patients with asthma potentially contribute to variability in asthma phenotype has not been studied. To explore this, we conducted a pilot study analysing 58 faecal samples collected from asthmatic and healthy non-asthmatic adults enrolled in a prospective observational study. We hypothesised that features characteristic of asthma or linked to asthma phenotype are associated with differences in faecal bacterial community profiles.
Methods
Clinical study
Subjects were enrolled in a prospective observational cohort study at the University of Michigan (Characterization of Adults for Asthma Microbiome Research Studies; NCT02887911). All subjects provided written informed consent. Subjects participated in two study visits which occurred within a 4-week period. At these characterisation visits, detailed assessments of asthma and allergy history were performed, including lung function testing (spirometry, methacholine challenge, bronchodilator reversibility) and asthma control (Asthma Control Questionnaire-710). Asthma diagnosis was confirmed by spirometry with methacholine challenge and/or bronchodilator reversibility testing, performed according to the American Thoracic Society/European Respiratory Society guidelines.11 Healthy non-asthmatic subjects had no history of lung diseases or chronic symptoms suggestive of such, as well as with normal spirometry and no evidence of airway hyper-responsiveness (methacholine PC20 >16 mg/mL or PD20 >400 µg; PC20 or PD20, provocative concentration or dose resulting in 20% decline in FEV1) or bronchodilator reversibility. Key exclusion criteria included significant smoking history (>10 pack-years); acute lower respiratory illness, asthma exacerbation and/or systemic antibiotic use within 8 weeks of visit 1 or between visits; significant cardiovascular history or other chronic medical condition besides asthma requiring immunosuppressive therapy; and for subjects with asthma, an absence of asthma symptoms in the past 2 years. Prescribed asthma medications had to be stable for 30 days before visit 1 and between visits.
Blood was collected to determine atopic sensitisation status to common respiratory allergens, using a clinically approved panel performed by the University of Michigan clinical laboratory (specific IgE to a panel of 16 aeroallergens; Phadia ImmunoCAP). Presence of at least one positive specific IgE test on this panel was considered evidence of atopic sensitisation. Subjects also completed a dietary questionnaire (NutritionQuest Block 2014; Berkeley, California) to capture macronutrient and micronutrient intake. Induced sputum was collected by inhalation of 3% saline for 12 min.12 Faecal samples were collected by subjects on the day or within 7 days of a study visit using a commercial kit containing a DNA stabilisation reagent (OMR-200; DNA Genotek). The majority of subjects submitted paired faecal samples for analysis.
Sample processing and data analysis
Inflammatory mediators were measured in plasma and sputum using a Luminex instrument and xMAP multiplex assays (Milliplex Human High Sensitivity T Cell 21 Plex, Milliplex Human Metabolic Hormone Expanded, Milliplex Human Adipocyte and Milliplex Human Soluble Cytokine Receptor; MilliporeSigma). A total of 58 faecal samples from 33 subjects were analysed. A punch biopsy tool (4.0 mm; Integra Miltex) was used to produce uniform cores of frozen stool, and the total DNA was extracted using a cetyltrimethylammonium bromide buffer-based protocol coupled with bead-beating.13 16S ribosomal RNA gene sequencing (V4 region) was performed by the University of Michigan’s Microbiome Core on an Illumina MiSeq platform (MiSeq Reagent Kit V2), following previously published methods.14 Sequence data were processed in mothur and aligned using the Ribosomal Database Project database. Bacterial taxa comprising <0.1% of the total community were removed and operational taxonomic unit (OTU) counts converted to relative abundance for downstream analyses. Samples were compared using beta-diversity measures such as Bray-Curtis dissimilarity (BCdissim) or Unifrac distances. Non-parametric tests (Wilcoxon rank-sum or Kruskal-Wallis) were used where appropriate for between-group comparisons, and generalised linear models (GLM) based on a negative binomial distribution used to determine differentially abundant taxa between prespecified groups or clinical variables of interest. GLM is a generalisation of conventional linear regression that allows for non-normal error distribution models in data where non-linear relationships likely exist, such as count-based microbial compositional data. In the particular implementation used in this study (R package mvabund), the GLMs incorporated adjustment for multiple testing using a step-down resampling procedure as described in the source package documentation. To explore intrinsic patterns of microbiota variation among subjects with asthma, k-means clustering was applied and between-cluster differences in clinical and biological features compared. Analyses were performed in R using open-source packages such as vegan and mvabund or published python-based scripts. Sequence data are deposited under BioProject ID PRJNA474717.
Results
Subjects with asthma in this study overall had mild-moderate disease and generally well-controlled symptoms (table 1). Lung function and asthma control measures were similar between visits, congruent with clinical stability. Of subjects with asthma, 58% were taking either a low-dose or medium-dose inhaled corticosteroid. Asthmatic and healthy subjects did not significantly differ in age, gender or body mass index (BMI). Subjects with asthma in this cohort had predominantly non-eosinophilic airway inflammation, suggestive of low type 2 immune responses. Blood eosinophil counts were marginally higher in subjects with asthma (Wilcoxon rank-sum, p=0.06).
Characteristics of subjects
Paired faecal bacterial profiles show short-term stability and associate with key features of asthma
Within-subject faecal bacterial microbiota profiles between visits were highly similar, indicating short-term stability of the gut microbiome in the absence of changes in health status. This was evaluated by several approaches, including distance-based permutational multivariate analysis of variance (PERMANOVA)15 and testing of the homogeneity of group dispersions by visit16 (p>0.05; figure 1). Shannon diversity measures within subject were also strongly correlated between visits (mean r=0.93, p<0.05), as well as within-subject OTU relative abundances between visits (mean Spearman’s r, 0.83±0.06, p<0.01).
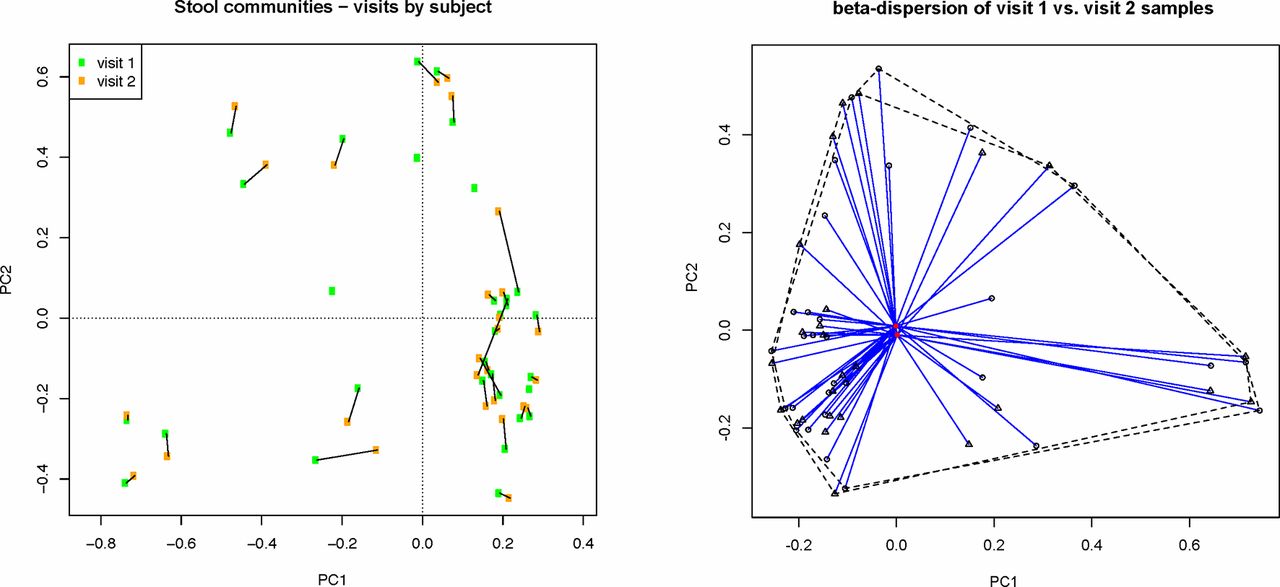
Paired samples from both visits display overall similarity in faecal bacterial community structure, as shown for paired samples by subject and testing of differences in group dispersions by visit based on Hellinger-transformed relative abundance data (p>0.05).
Leveraging the availability of paired gut microbiota data, BCdissim and Unifrac distance measures were calculated between visits and the values used as representative read-outs of each subject’s gut bacterial community. Using these measures, we explored whether variation in gut microbiota composition between subjects related to key features of asthma. The degree of sensitisation to respiratory allergens was inversely correlated with BCdissim (number of positive specific IgE tests; Spearman’s r=–0.64, p=0.02; figure 2A), suggesting that differences in gut microbiota composition are associated with the degree of aeroallergen sensitisation and with asthma since nearly all subjects with asthma were sensitised in this cohort. Additionally, lung function (forced expiratory volume in 1 s (FEV1) %predicted at visit 1) was inversely correlated with unweighted Unifrac distances (r=–0.48, p=0.01; figure 2B). Using loading scores from principal component analysis of Hellinger-transformed relative abundance data, FEV1 %predicted was most strongly associated with variation in faecal bacterial community structure along principal component 2 (PC2) (figure 2C; r=0.50, p=0.02, repeated-measures correlation).17 Collectively, the results from these independent analytic approaches suggest significant relationships between gut bacterial community structure, greater aeroallergen sensitisation and decreased lung function, the latter two being hallmarks of allergic asthma.
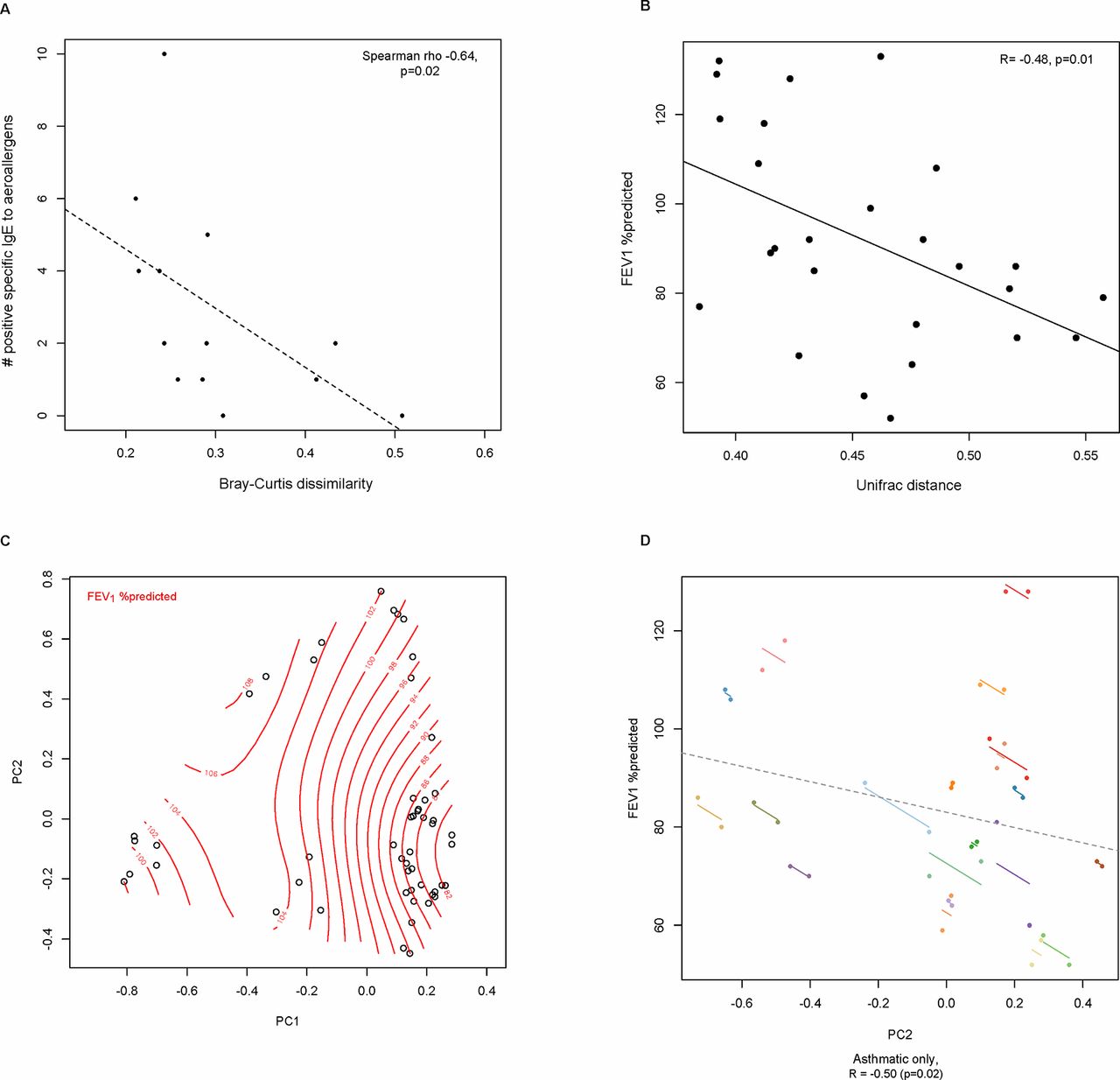
Differences in gut bacterial microbiota composition are associated with (A) degree of aeroallergen sensitisation; (B) lung function as measured by FEV1 %predicted (unweighted Unifrac); and (C) Principal component analysis of bacterial relative abundance data (Hellinger-transformed) of all available samples, with splines representing FEV1 %predicted values. (D) Differences in FEV1 %predicted correlate most strongly with gut bacterial compositional variation along principal component 2 (PC2 loading scores of paired asthmatic samples coloured by subject; repeated-measures correlation). FEV1, forced expiratory volume in 1 s; PC1, principal component 1; PC2, principal component 2.
Differences in specific gut bacterial communities associate with asthma and asthma phenotype
Given the similarity of gut microbiota profiles between visits, further analyses were performed using only visit 1 data, which allowed for application of statistical methods that assume independent observations. The results of GLMs (mvabund, negative binomial distribution) confirmed a strong association between FEV1 and differences in gut bacterial composition most evident at the phylum level, in particular Bacteroidetes and Firmicutes. Notably, the association with FEV1 remained statistically significant even after single-variable adjustment in separate models for age, gender or BMI (Bacteroidetes, padj=0.002; Firmicutes, padj=0.003). The ratio of Bacteroidetes/Firmicutes relative abundance was lower in subjects with asthma (median ratios: 0.42 vs 0.55; online supplementary figure S1).
Supplemental material
Reflective of asthma’s heterogeneity, phenotyping schemes in prior studies have incorporated various types of features, including clinical data, physiological measures, and inflammatory or molecular biomarkers.18 To incorporate gut microbiota data and begin to explore potential relationships to asthma phenotype, we pursued two approaches. First, we applied a feature selection approach,19 where each feature is tested on its independent ability to classify asthmatic versus healthy status. Only the top 60 OTUs by rank abundance were included. Gut microbiota members with the highest F-scores included OTUs that classified to Bacteroides (F-score 0.34), Enterobacteriaceae family (0.32), Bifidobacterium (0.11) and Lachnospiraceae family (0.11). Relative abundances of the first two were decreased in subjects with asthma (eg, Bacteroides, 6% vs 12%), while relative abundances of the latter two were increased in the asthmatic group (eg, Bifidobacterium, 4% vs 1%). These observations support the potential value of incorporating gut microbiota data in future strategies to better understand asthmatic phenotype.
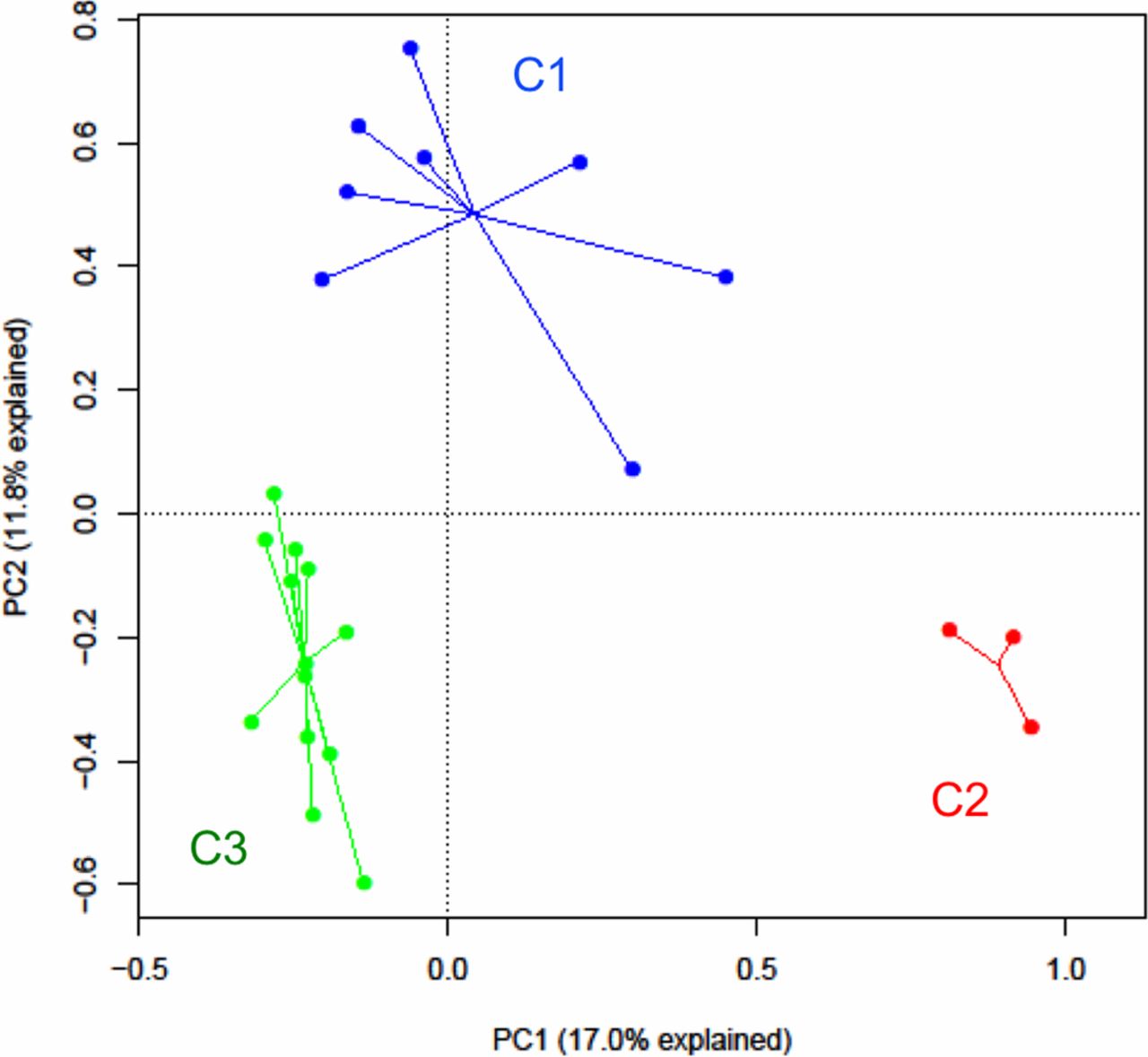
Next, to explore intrinsic patterns of variation in gut bacterial community structure among subjects with asthma only, k-means clustering was performed, which revealed three distinct clusters (C1, C2, C3; figure 3). The majority segregated into C1 or C3 (n=8 and 13 subjects, respectively). Testing of related metadata revealed differences between the clusters in several clinical and inflammatory features of interest (table 2). Most notable was a significant difference in lung function, with the lowest lung function (FEV1) observed in C3 subjects. This was paralleled by differences between the clusters in bronchial hyper-responsiveness (eg, PC20) and bronchodilator reversibility. Clear trends were evident even though they did not reach statistical significance. In particular, C3 subjects had greater airway hyper-responsiveness (PC20) and bronchodilator reversibility than C1 subjects. In contrast, C1 subjects had more normal lung function, and the majority of obese subjects with asthma in this study belonged to C1. C1 subjects also displayed less airway hyper-responsiveness, less bronchodilator reversibility and had the lowest forced vital capacity. C2 was the smallest cluster with similar lung function as C3 but with a distinct gut bacterial composition characterised by lowest community diversity and high Prevotella abundance. The stability of the clustering pattern was assessed by iteratively removing individual samples, as well as all three in C2, and reperforming the k-means clustering. The cluster assignment of the remaining samples in each iteration overall did not significantly change, suggesting robust differences in gut bacterial community structure in this group of subjects with asthma. Distance-based PERMANOVA testing also independently confirmed a significant association between cluster assignment (C1, C2, C3) and compositional variation among samples (p<0.01). Age, gender distribution and dietary factors except daily fruit intake did not significantly differ between these clusters. We also observed trends towards differences in inflammatory mediators such as blood soluble receptor for advanced glycation end-products (sRAGE; p=0.08) and sputum interleukin (IL)-8 (p=0.09) and IL-1β (p=0.10). These findings suggest links between differences in gut microbiome patterns and markers of asthma severity (FEV1, bronchial hyper-responsiveness) as well as inflammatory features that may prove important to asthmatic phenotype.
{kind=link}
{kind=link}
![[bmjresp-2018-000324-supp1.png]](https://bmjopenrespres.bmj.com/content/bmjresp/5/1/e000324/DC1/embed/inline-supplementary-material-1.png?download=true){kind=link}
{kind=link}
k-Means clustering of gut bacterial compositional profiles in subjects with asthma. Clusters differ by several clinical features including lung function, as shown in table 2. PC1, principal component 1; PC, principal component 2.
Three gut bacterial community clusters in the asthmatic group differ in several clinical and inflammatory features (median values unless otherwise noted)
Discussion
This pilot study aimed to explore the relationships between gut bacterial microbiota composition and features of asthma in a well-characterised adult cohort recruited at a single institution. This work represents, to our knowledge, the first detailed exploration of gut microbiota relationships to asthmatic features in adults with established disease. The topic is highly relevant as adult asthma is phenotypically heterogeneous, linked to differences in airway inflammation patterns as well as systemic inflammation,20 21 and associated with differences in treatment responses. The mechanistic underpinnings of many asthma phenotypes remain poorly understood.
The results of this study provide the first evidence that differences in gut bacterial microbiota composition associate with differences in lung function, along with other phenotypic features of asthma. Associations with lung function were consistent across different independent methods of analysing the gut microbiota data and remained significant even after adjusting in GLMs for age, gender or BMI. This suggests a robust association between variation in the gut microbiome and lung function, further highlighted by the results of our cluster analysis in the asthmatic group alone. The significant differences in lung function between the asthma-associated gut microbiota clusters were striking, as other characteristics like age, gender and macronutrient intake patterns did not differ. The latter is likely reflective of the fairly homogeneous subject demographics in this pilot study. Moreover, the differences were not driven solely by obese subjects who may define cluster 1 but not the other two asthmatic clusters in whom lung function was actually much lower. Cluster 2 was very small, but intriguingly Prevotellaceae members were the most abundantly represented. Prevotella-dominated gut microbiota patterns and specific Prevotella species have been associated with chronic inflammatory diseases.22 Overall, our findings suggest potential gut-derived factors or mediators of inflammatory immune responses that may play a role in specific phenotypes of asthma, which require further study. Such studies should aim to understand the likely bidirectional interactions between gut microbiota and the immune system that may contribute to asthma heterogeneity in adults.
This pilot study has both limitations and strengths. First, being a pilot study, the overall number of unique subjects was small, but we had a very high rate of return of stool samples and nearly 90% of subjects submitted paired samples. This allowed for assessment of short-term stability in gut microbiota patterns, an improvement on single time-point assessments that helps to establish the baseline characteristics of a subject’s microbiome during clinical stability and facilitate interpretation of results in longitudinal assessments. Second, subjects were extensively characterised in our study protocol using multiple tools to capture features relevant to asthma phenotype, including formal lung function testing to confirm or rule out asthma. We also evaluated aeroallergen sensitisation by serological testing, and interestingly a prior study reported increased prevalence of histamine-secreting microbes in adult subjects with asthma.23 Although approximately half of the asthmatic group were taking an inhaled corticosteroid as chronic therapy, we did not find statistically significant associations between this treatment and gut bacterial composition which might relate to the small sample size. The study was conducted at a single academic institution located in the upper Midwestern region of the USA. Thus, demographic and other characteristics of subjects analysed in this study are relatively more homogeneous, which may account for the lack of observed associations with dietary intake, variations in which are associated with differences in gut microbiota composition.24 Conversely, this likely strengthens the importance of our findings.
In summary, this is the first study to demonstrate a significant relationship between gut microbial community structure, aeroallergen sensitisation and lung function in a group of well-characterised asthmatic and non-asthmatic adults. This suggests potentially important relationships between gut microbiota functions and asthma in established disease, but further studies are needed to validate this in larger cohorts and determine the mechanistic pathways involved. Investigations that build on these 16S ribosomal RNA gene-based findings and incorporate other tools to understand functions of and mediators derived from the gut microbiota will be necessary to generate further insight into these microbiome relationships to adult asthma phenotype.
Acknowledgments
We thank Liujian (Jane) Zhao for her extensive technical support of the CAARS study, including processing of stool samples for this analysis.
References
Footnotes
Contributors Study design: YJH, KR. Subject recruitment, sample processing, clinical data collection and analysis: LB, AB, ON, KR, YJH. Microbiota data analysis: LB, SM, KO, JRE-D, YJH. Data interpretation and manuscript writing/review: all authors.
Funding Research supported by the NIAID (R01AI129958) (YJH), the University of Michigan Host-Microbiome Initiative, and the Michigan Institute for Clinical and Health Research (UL1TR000433).
Competing interests None declared.
Patient consent Not required.
Ethics approval The study was approved by the University of Michigan Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Sequence data are deposited under BioProject ID PRJNA474717.