Article Text

Abstract
Introduction Idiopathic pulmonary fibrosis (IPF) greatly impacts quality of life and eventually leads to premature death from respiratory failure. Inhaled treprostinil was associated with improvements in forced vital capacity (FVC) and reduced exacerbations of underlying lung disease in post hoc analyses from a phase 3 study in patients with precapillary pulmonary hypertension due to interstitial lung disease. These results, combined with preclinical evidence of treprostinil’s antifibrotic activity, support its investigation in the treatment of IPF.
Methods and analysis The TETON programme consists of two replicate, 52-week, randomised, double-blind placebo-controlled, phase 3 studies, each enrolling 396 subjects (NCT04708782, NCT05255991). Eligible subjects must have a diagnosis of IPF confirmed by central imaging review, along with an FVC ≥45%. Stable background use of pirfenidone or nintedanib is allowed. The primary endpoint is change in absolute FVC at week 52. Secondary endpoints include time to clinical worsening (first event of death, respiratory hospitalisation or ≥10% decline in % predicted FVC), time to first acute exacerbation of IPF, overall survival, change in % predicted FVC and change in the King’s Brief Interstitial Lung Disease Questionnaire at week 52. Safety parameters include adverse events, hospitalisations, oxygenation and laboratory parameters. Patients who complete week 52 will be eligible to enter an open-label extension study.
Ethics and dissemination Studies will be conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice, Declaration of Helsinki principles, and local regulatory, ethical and legal requirements. Results will be published in a peer-reviewed publication.
- Interstitial Fibrosis
- Nebuliser therapy
- Rare lung diseases
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. No data are available. No data haVE been generated/analySed for these studies as of yet. This submission is a prospective publication of the study design only.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Idiopathic pulmonary fibrosis (IPF) is a serious, chronic, progressive, fibrosing interstitial pneumonia with no known cause. Two antifibrotic therapies, nintedanib and pirfenidone, are approved for treatment of IPF, but additional therapies are necessary to further reduce morbidity and improve survival. In a post hoc analysis of the INCREASE study, inhaled treprostinil significantly improved forced vital capacity in subjects with pulmonary hypertension due to interstitial lung disease, with greatest improvement in subjects with IPF.
WHAT THIS STUDY ADDS
Two replicate, randomised, placebo-controlled studies, TETON 1 and 2, will evaluate the safety and efficacy of inhaled treprostinil for treatment of IPF for 52 weeks.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The prospectively designed TETON clinical trial programme currently underway aims to definitively confirm whether inhaled treprostinil has antifibrotic effects and may offer a much needed treatment option for this vulnerable group of patients.
Introduction
Idiopathic pulmonary fibrosis (IPF), a distinct type of interstitial lung disease (ILD), is a serious, chronic, progressive, fibrosing interstitial pneumonia with no known cause that typically occurs in patients above 50 years of age.1. It is characterised by progressive fibrosis, and a radiological and/or histopathological pattern known as usual interstitial pneumonia (UIP) in the absence of an alternate aetiology for this pattern.2 UIP usually presents as ‘honeycombing’ (subpleural cystic airspaces with well-defined walls), traction bronchiectasis (dilatation of the bronchi) and peripheral alveolar septal thickening. IPF is associated with increasing cough and dyspnoea, greatly impacts patients’ quality of life and eventually leads to premature death from respiratory failure or complicating comorbidities. Diagnosis is based on identification of the UIP pattern on high-resolution CT (HRCT) scan and/or lung biopsy and exclusion of other ILDs/similar conditions.1 3 Only two therapies are currently approved for IPF, nintedanib and pirfenidone. Nintedanib is a kinase inhibitor approved for the treatment of IPF, chronic fibrosing ILDs with a progressive phenotype, and systemic sclerosis-associated ILD.4 Pirfenidone belongs to the pyridone class and is approved for the treatment of IPF.5
Treprostinil is a stable analogue of prostacyclin, which promotes vasodilation of pulmonary and systemic arterial vascular beds and inhibits platelet aggregation.6 The inhaled formulation of treprostinil is approved in the USA, Argentina and Israel for the treatment of WHO group 1 and in the US for WHO Group 3 (PH-ILD) pulmonary hypertension.7–9 In addition to its effects on the pulmonary vasculature, there are data to suggest that treprostinil has antifibrotic properties. Specifically, treprostinil has been shown to have antifibrotic effects via several mechanisms in vitro:
Dose-dependent prevention of fibroblast proliferation to decrease extracellular matrix composition via a TGF-beta1 and PDGF-BB antagonism in human peripheral lung fibroblasts.10
Inhibition of extracellular matrix-deposition by fibroblasts by both cyclic adenosine monophosphate (cAMP)-dependent and non-dependent mechanisms in human peripheral lung fibroblasts.11
Suppression of profibrotic fibroblast activity and the synthesis and deposition of collagen and fibronectin in mice, with anti-inflammatory processes mediated by NK-kappaB signalling.12 13
Suppression of profibrotic fibroblast activity through Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) inhibition from prostacyclin (IP) receptor activation.14
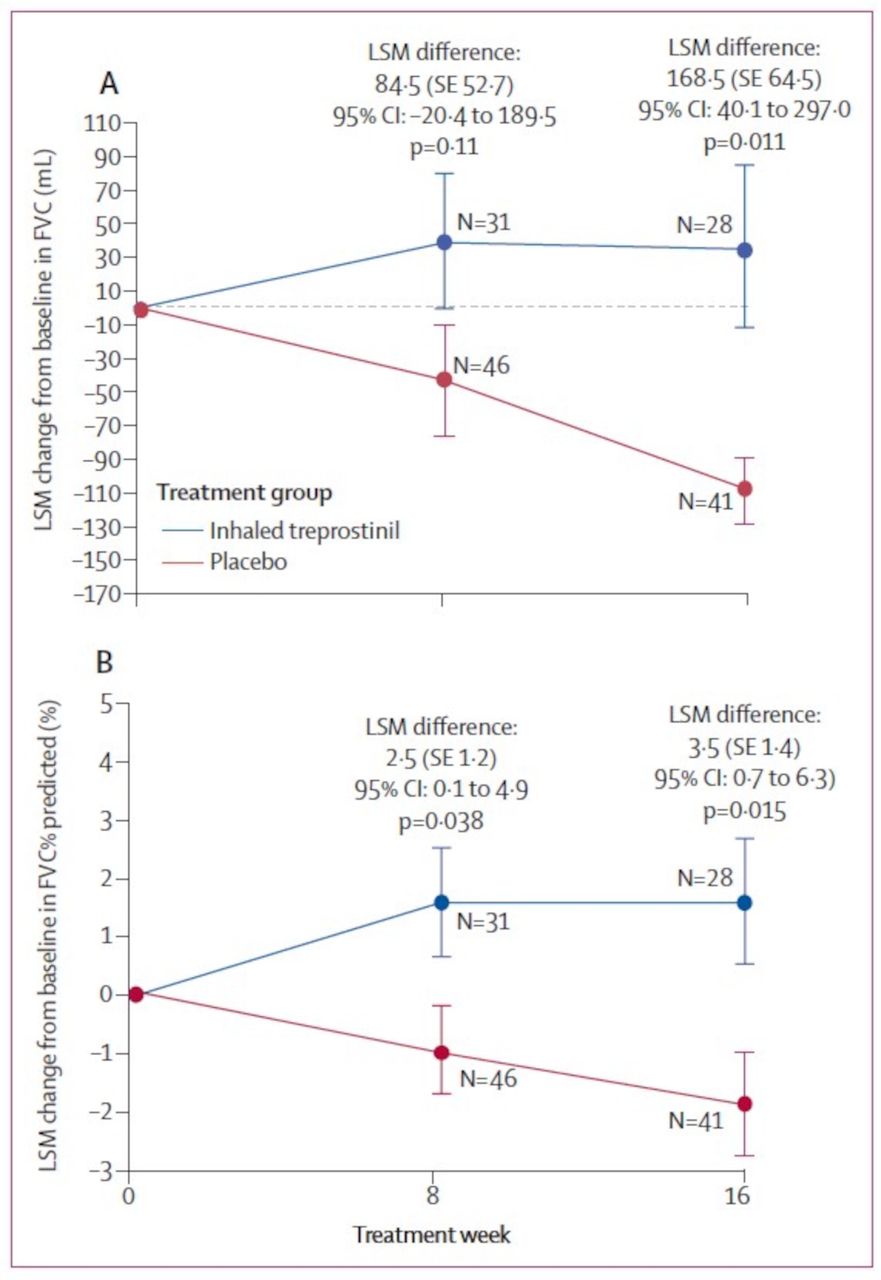
The results of the INCREASE study served as the basis for the WHO Group 3 (PH-ILD) indication for inhaled treprostinil within the USA and has also provided robust evidence that inhaled treprostinil may benefit IPF patients.8 The INCREASE study was a multicentre, randomised, double-blind, placebo controlled, 16-week, parallel group study, designed to investigate the safety and efficacy of inhaled treprostinil in 326 subjects with PH-ILD, including PH associated with IPF. In addition to meeting the primary (6-minute walk distance (6MWD)) and secondary endpoints, post hoc analysis of the INCREASE study demonstrated that inhaled treprostinil resulted in significant improvements in forced vital capacity (FVC) in subjects with PH-ILD.15 Specific analysis of patients with IPF showed FVC differences of 84.5 mL (SE 52.7; 95% CI –20.4 to 189.5; p=0.11) at week 8 and 168.5 mL (SE 64.5; 95% CI 40.1 to 297.0; p=0.011) at week 16 (figure 1). Additionally, there were significantly fewer lung disease exacerbations in the treprostinil group than the placebo group. Specifically, 43 (26%) of 163 patients in the treatment group had an exacerbation of underlying lung disease compared with 63 (39%) of 163 patients in the placebo group (p=0.02 by Fisher’s exact test).15 In the 108-week open-label extension of the INCREASE study, these notable improvements in FVC were sustained for patients randomised to active treatment, and marked improvements were shown for those subjects formerly assigned to placebo once starting inhaled treprostinil.16

Change in FVC at weeks 8 and 16 for patients with idiopathic pulmonary fibrosis in the increase study. FVC, forced vital capacity. LSM, least-squares mean
The improvements in FVC and reduced exacerbations of underlying lung disease from the INCREASE study, combined with the preclinical evidence of antifibrotic activity of treprostinil, suggest that inhaled treprostinil may offer a treatment option for patients with IPF. TETON will be the first clinical programme of an inhaled therapy for IPF.
Methods
Study design
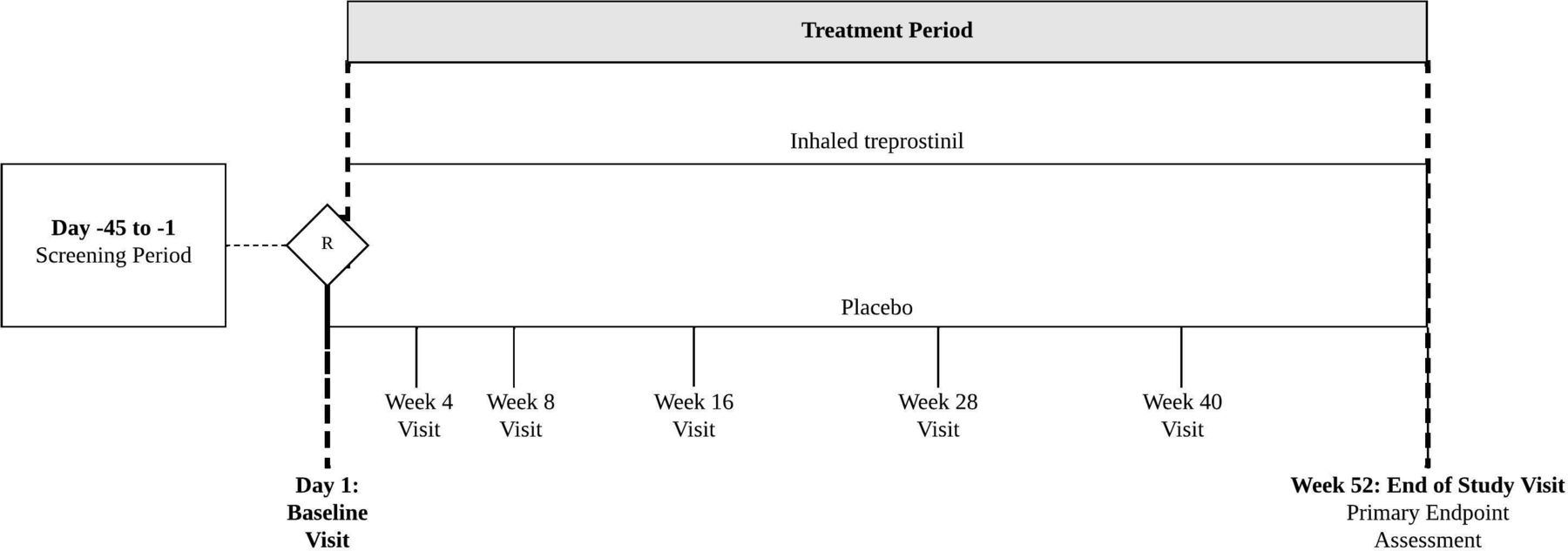
The TETON programme consists of two, 52-week, randomised, double-blind placebo-controlled, phase 3 studies with nebuliser solution treprostinil (Tyvaso) in patients with IPF (figure 2). Each study will enrol approximately 396 subjects. Subjects who complete the randomised studies may be eligible to rollover into an open-label extension study assessing long-term safety and tolerability of inhaled treprostinil.
{kind=link}
{kind=link}
TETON phase 3 study designs.
Study population
Subject eligibility will be based on inclusion and exclusion criteria described in boxes 1 and 2 below and includes subjects with a diagnosis of IPF based on the 2018 American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Latin American Thoracic Society (ALAT) Clinical Practice Guideline and confirmed by central review of HRCT imaging.1 Approximately 396 eligible subjects will be randomly assigned 1:1 in each study to nebuliser solution treprostinil (Tyvaso) or placebo at Baseline. Randomisation will be stratified by the use of IPF background therapy (nintedanib or pirfenidone vs no background therapy). All subjects will initiate inhaled treprostinil (6 μg/breath) or placebo at a dose of 3 breaths (18 μg) administered four times daily (during waking hours) and will titrate to a target dosing regimen of 12 breaths (72 μg) four times daily.
Key inclusion criteria for phase 3 randomised TETON studies
Key inclusion criteria
Age: ≥40 years old.
Forced vital capacity ≥45% predicted.
If on background pirfenidone or nintedanib for idiopathic pulmonary fibrosis (IPF), stable dose for ≥30 days prior to baseline.
Diagnosis of IPF based on the 2018 ATS/ERS/JRS/ALAT Clinical Practice Guidelines.1
High-resolution CT within the last 12 months ‘consistent with usual interstitial pneumonia’ (confirmed by central reader).
Females of childbearing potential: must have confirmed negative pregnancy test at screening and baseline and willing to practice abstinence or use 2 forms of birth control for duration of study and 30 days after discontinuing study drug.
Males with a partner of childbearing potential must use a condom for the duration of treatment and for at least 48 hours after discontinuing study drug.
Able to communicate effectively with study personnel, and is considered reliable, willing and likely to be cooperative with protocol requirements.
Key exclusion criteria for phase 3 randomised TETON studies
Key exclusion criteria
Pregnant or lactating.
Primary obstructive disease: prebronchodilator forced expiratory volume in 1 second/forced vital capacity<0.70.
Diagnosis of connective tissue disease-interstitial lung disease or combined pulmonary fibrosis and emphysema.
More than 10 L/min of supplemental oxygen at rest at baseline.
Receipt of any prostacyclin therapy, IP receptor agonist, endothelin-receptor antagonist, or phosphodiesterase-5 inhibitor or soluble guanylate cyclase stimulator within 60 days prior to baseline (washout permitted).
Shown intolerance or significant lack of efficacy to a prostacyclin or prostacyclin analogue that resulted in discontinuation or inability to effectively titrate therapy
Myocardial infarction within 6 months prior to baseline or unstable angina within 30 days prior to baseline.
Use of any of the following medications:
Azathioprine (AZA), cyclosporine, mycophenolate mofetil, tacrolimus, oral corticosteroids (OCS)>20 mg/day or the combination of OCS+AZA+N-acetylcysteine within 30 days prior to baseline.
Cyclophosphamide within 60 days prior to baseline.
Rituximab within 6 months prior to baseline.
Exacerbation of idiopathic pulmonary fibrosis (IPF) or active pulmonary or upper respiratory infection within 30 days prior to baseline.
Antibiotic or steroid regimens for infection or acute exacerbation must be completed more than 30 days prior to baseline.
Must have been discharged more than 90 days prior to baseline ifhospitalised for an acute exacerbation of IPF or a pulmonary or upper respiratory infection.
Any condition that would interfere with the interpretation of study assessments or would impair study participation or cooperation.
Objectives and endpoints
The primary efficacy endpoint of the 52-week studies is the change in absolute FVC in subjects with IPF from baseline to week 52 (box 3). Spirometry measures will be centrally reviewed to confirm eligibility at Screening and to ensure quality of the primary endpoint. FVC is considered the gold-standard efficacy endpoint for IPF clinical studies and was used to gain approval of both nintedanib and pirfenidone globally for treatment of IPF.
Study endpoints
Primary endpoint:
Change in absolute forced vital capacity (FVC) in subjects with idiopathic pulmonary fibrosis (IPF) from baseline to week 52.
Secondary endpoints:
Time to clinical worsening (including time to death, respiratory hospitalisation or ≥10% relative decline in % predicted FVC).
Time to first acute exacerbation of IPF.
Overall survival at week 52.
Change from baseline in % predicted FVC at week 52.
Change from baseline in King’s Brief Interstitial Lung Disease Questionnaire score at week 52.
The secondary endpoints in these studies will include time to clinical worsening, time to first acute exacerbation of IPF, overall survival at week 52, change from baseline in % predicted FVC at week 52, and change from baseline in King’s Brief Interstitial Lung Disease Questionnaire score at week 52 (box 3).
An independent Clinical Endpoint Committee (CEC) will be used during this study to adjudicate respiratory-related hospitalisations, acute exacerbations of IPF and deaths on an ongoing basis. The CEC will be composed of clinicians who are not participating study Investigators. The independent CEC will review and confirm all specified events in a blinded fashion throughout the conduct of the study in accordance with the established charter.
Sample size
A sample size of 336 subjects will have at least 80% power to detect a treatment difference in means of 95 mL in change from baseline in absolute FVC, assuming that the common SD is 300 mL using a two-group t-test with a 0.05 two-sided significance level. Assuming a 15% drop-out rate, 396 subjects need to be randomised in each trial. The assumptions related to sample size may be evaluated by review of blinded FVC data during the study and, if necessary, sample size will be recalculated.
Results will be published in a peer-reviewed publication. Patients/the public were not involved in the design, conduct, reporting or dissemination plans of the studies.
Informed consent
All prospective subjects will give informed consent prior to the conduct of any study-related procedures.
Study governance
Study steering committee members, along with the sponsor, contributed to the design of these studies. The Steering Committee will provide ongoing oversight of the programme, including study conduct, review of output from the data monitoring committee (DMC) when necessary, and input on study results and data analysis.
An independent external DMC is established for the programme and includes physicians knowledgeable in the treatment of IPF. Throughout the course of the studies, the DMC will meet on a regular basis to monitor safety and will be unblinded to individual subject treatment allocation during the review process. The sponsor and the study team will not have access to unblinded study data during this process.
Discussion
Despite the approval of two antifibrotic therapies, nintedanib and pirfenidone, IPF mortality remains high and patients’ quality of life remains low. Neither therapy has been shown to improve FVC or improve quality of life, but simply slow disease progression. In addition to showing significant improvement in 6MWD the clinical data from subjects in the INCREASE study with both PH and IPF demonstrated statistically significant improvements in FVC over 16 weeks, suggesting that inhaled treprostinil may have antifibrotic properties in addition to vasodilatory properties. The TETON clinical programme aims to determine whether inhaled treprostinil may provide significant benefit for patients with IPF alone, agnostic of the presence of pulmonary hypertension, over a longer study duration of 52 weeks compared with the INCREASE study’s 16-week duration.
TETON is the first clinical programme of an inhaled therapy for IPF. It is possible that direct administration to the lung may confer additional benefit for treatment with potentially fewer adverse effects compared with systemically delivered therapies. The TETON clinical programme aims to study inhaled treprostinil in a setting that mimics real-world treatment for IPF by not unnecessarily restricting the inclusion and exclusion criteria compared with other studies for IPF. Notably, there is no upper age limit, subjects on the lung transplant list are not excluded from participation, and FVC must be ≥45% (no upper cap). Patients may be on background pirfenidone or nintedanib therapy provided they are on a stable and optimised dose for at least 30 days prior to baseline. Criteria to exclude only the sickest subjects (life expectancy <6 months, IPF exacerbation within 30 days prior to baseline, respiratory-related hospitalisation within 90 days prior to baseline) are specified to decrease risks of data loss due to drop out since the study is 52 weeks long.
The primary efficacy endpoint of TETON 1 and 2 is the change in absolute FVC from baseline to week 52. FVC is considered the gold-standard efficacy endpoint for IPF clinical studies and has been used to gain approval of both nintedanib and pirfenidone globally for treatment of IPF. FVC is considered to be a reliable, valid and responsive endpoint linked to patient-sensitive outcomes like dyspnoea and mortality .17 FVC assesses and reflects changes in disease burden in patients with fibrotic lung diseases, and measured declines indicate disease progression. A 10% decline in FVC over 12 months is felt to be clinically relevant and is associated with increased mortality.18 Additional clinically relevant endpoints, including externally adjudicated exacerbations, hospitalisations and mortality events, will be collected to assess the overall safety and efficacy of inhaled treprostinil for the treatment of IPF. To protect the integrity and quality of the primary endpoint (FVC) and to reduce overall operational burden on sites and subjects, the 6MWT was not included in the TETON clinical study. The 6MWT results from the INCREASE study have already demonstrated the benefits of inhaled treprostinil on exercise capacity.
Conclusion
The clinical results from the INCREASE study appear to demonstrate a beneficial effect on loss of lung function in patients with ILD, including IPF, and associated pulmonary hypertension. These findings, along with the preclinical evidence of antifibrotic activity of treprostinil, are intriguing and hypothesis generating. The prospectively designed TETON clinical trial programme currently underway aims to definitively confirm this intriguing hypothesis and may offer a much needed treatment option for this vulnerable group of patients.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. No data are available. No data haVE been generated/analySed for these studies as of yet. This submission is a prospective publication of the study design only.
Ethics statements
Patient consent for publication
Ethics approval
Studies will be conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice, Declaration of Helsinki principles, and local regulatory, ethical, and legal requirements.
Footnotes
Contributors PS, CQD, NP, HB and LP are involved in the study design, conduct, oversight and submission of the TETON clinical programme as employees of United Therapeutics. SDN, JB, VC, LL and KRF are involved in the design of the TETON clinical programme and provide external oversight and consulting for the programme as members of the Steering Committee. SDN, JB, VC, LL, PS, CQD, NP, HB, LP and KRF were involved in the authoring, review and approval of the draft and final manuscripts.
Funding The TETON clinical programme and this manuscript is funded by United Therapeutics Corp. (Research Triangle Park, NC).
Competing interests SDN has received personal fees from United Therapeutics for consulting services and speaking arrangements. JB has received personal fees from Boehringer Ingelheim, Biogen, BMS, Roche, Sanofi/Genzyme, AstraZeneca, Roche, Novartis and United Therapeutics; support for attending meetings/travel from Boehringer-Ingelheim; personal fees for participation on a data safety monitoring board or Advisory Board for Actelion; and is the head of the German ILD Guideline Committee. VC has received grants or contracts from Boehringer Ingelheim;. personal fees from Boehringer Ingelheim, Roche, Galapagos, Sionogi, RedX, Pure Tech, Celgene/BMS, AstraZeneca, CSL Behring, and Sanofi; support for attending meetings/travel from Boehringer Ingelheim and Roche; participates on a data safety monitoring board for Roche, Galapagos and Galecto; and is on an Adjudication Committee for Fibrogen. LL has received research grants from Boehringer Ingelheim, Novartis, Celgene, Biogen, Respivant, Galapagos, Galecto, Roche, Fibrogen, Pliant, Genentech and Veracyte; personal fees from AstraZeneca, BMS, United Therapeutics, Daewong, Trevi, Pliant, Veracyte, DevPro, Genentech, and Boehringer Ingelheim; is on an advisory board for DevProBiopharma; and is on the Pulmonary Fibrosis Foundation Registry Steering Committee. SDN, JB, LL, VC and KRF are on the Steering Committee for the TETON programme. PS, CQD, NP, HB and LP are employees of United Therapeutics and have received salaries and stock options as part of their employment. KRF has received personal fees from Boehringer Ingelheim, Roche/Genentech, Bellerophon, Respivant, Shionogi, DevPro, AstraZeneca, Pure Health, Horizon, Fibrogen, Sun Pharmaceuticals, Pliant, United Therapeutics, Arrowhead, Lupin, Polarean, PureTech, Trevi Pharmaceuticals, CSL Behring, Daewong, Dispersol, Immumet, and NeRRe Therapeutics; has received grants or contracts from Boeringher Ingelheim; and royalties/licenses from UpToDate.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.