Article Text

Abstract
Introduction Antisynthetase syndrome (ASyS) is a rare autoimmune connective tissue disease (CTD), associated with autoantibodies targeting tRNA synthetase enzymes, that can present to respiratory (interstitial lung disease (ILD)) or rheumatology (myositis, inflammatory arthritis and systemic features) services. The therapeutic management of CTD-associated ILD and idiopathic pulmonary fibrosis (IPF) differs widely, thus accurate diagnosis is essential.
Methods We undertook a retrospective, multicentre observational cohort study designed to (1) evaluate differences between ASyS-associated ILD with IPF, (2) phenotypic differences in patients with ASyS-ILD presenting to respiratory versus rheumatology services, (3) differences in outcomes between ASySassociated with Jo-1 versus non-Jo-1 autoantibodies and (4) compare long-term outcomes between these groups.
Results We identified 76 patients with ASyS-ILD and 78 with IPF. Patients with ASyS were younger at presentation (57 vs 77 years, p<0.001) with a female predominance (57% vs 33%, p=0.006) compared with IPF. Cytoplasmic staining on indirect immunofluorescence was a differentiating factor between ASyS and IPF (71% vs 0%, p<0.0001). Patients with ASyS presenting initially to respiratory services (n=52) had a higher prevalence of ASyS non-Jo-1 antibodies and significantly fewer musculoskeletal symptoms/biochemical evidence of myositis, compared with those presenting to rheumatology services (p<0.05), although lung physiology was similar in both groups. There were no differences in high-resolution CT appearances or outcomes in those with Jo-1 versus non-Jo-1 ASyS-ILD.
Conclusions Extended autoimmune serology is needed to evaluate for ASyS autoantibodies in patients presenting with ILD, particularly in younger female patients. Musculoskeletal involvement is common in ASyS (typically Jo-1 autoantibodies) presenting to rheumatology but the burden of ILD is similar to those presenting to respiratory medicine.
- interstitial fibrosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
What are the key clinicoserological differences between antisynthetase syndrome (ASys)-associated lung disease presenting to respiratory services and (1) idiopathic pulmonary fibrosis (IPF) or (2) ASys presenting to rheumatology services?
What is the bottom line?
Patients with ASyS-associated interstitial lung disease (ILD) presenting to respiratory services are less likely to have overt myopathy at presentation than those presenting to rheumatology services. The presence of an anti-cytoplasmic stain on indirect immunofluorescence should alert clinicians to the possibility of ASyS (specifically if anti-nuclear antibody negative).
Why read on?
ASys-associated ILD is an under-recognised form of autoimmune-associated ILD that has different treatment pathways and better outcomes than IPF.
Introduction
The interstitial lung diseases (ILD) are a heterogeneous group of lung diseases with varying degrees of inflammation and/or fibrosis.1 Anti-synthetase syndrome (ASyS) is a rare autoimmune connective tissue disease (CTD) associated with autoantibodies targeting cytoplasmic tRNA synthetase enzymes. The exact role of these antibodies in disease pathogenesis remains to be elucidated. Clinical features typically include inflammatory myopathy, inflammatory arthritis, mechanic’s hands (thickened, hyperkeratotic and fissured aspects of the radial sides of the fingers) and ILD, while the presence of Raynaud’s phenomenon, Gottron’s papules, sicca symptoms and fever are useful ancillary features.2 In some patients, musculoskeletal manifestations may be absent or subclinical and mild.
To date, eight ASyS antibodies have been clinically described, although much of the existing literature reports on the manifestation of anti-Jo-1 disease, the most common of the anti-ASyS antibodies and directed against histidyl t-RNA synthetase. Others include anti-PL7, anti PL-12, anti-OJ, anti-EJ, anti-KS, anti-Ha, anti-Zo and generally occur mutually exclusive of each other.3 Current literature conflicts as to whether the antibody specificity influences outcomes in these patients.2 4–6 ILD is the most common extramusculoskeletal manifestation, with a prevalence ranging from 67% to 100%4; it may be presenting manifestation of ASyS or dominant feature: ‘lung dominant ASyS’. The long-term prognosis of ASyS has shown to be less severe than idiopathic pulmonary fibrosis (IPF) and importantly the therapeutic management of CTD-associated ILD and IPF differs widely, thus accurate diagnosis is essential.
The gold standard for autoantibody detection is immunoprecipitation (IPP), which is labour-intensive, expensive and restricted to specialised reference laboratories. Moreover, IPP analysis can take several weeks and so may not be suitable for clinical practice, where positive or negative results can influence clinical management. Anti-Jo-1 antibodies can be tested on most extractable nuclear antigen (ENA) assays (immunodiffusion or ELISA). The emergence of commercially available solid-phase immunoassays has facilitated more widespread assessment of other ASyS autoantibodies, better equipping clinicians with tools to support early diagnosis and appropriate management.
The aim of this multicentre study was to (1) evaluate differences between ASyS-associated ILD with IPF, (2) examine phenotypic differences in patients with ASyS-ILD presenting to respiratory versus rheumatology services, (3) examine differences in outcomes between ASyS associated with anti-Jo-1 versus non-Jo-1 autoantibodies and (4) compare long-term outcomes within these patient groups.
Methods
Study design
This was a retrospective, multicentre observational cohort study undertaken across three tertiary referral centres in the UK (Bristol, Liverpool and Bath) providing combined specialist rheumatological and ILD services. The study was approved by the Health Research Authority and health and Care Research Wales (HCRW), UK (IRAS 234299) and local R&D approval obtained at each site. In view of the retrospective study design, using data collected as part of routine clinical care, it was determined that written patient consent was not required.
Study subjects
Consecutive adult patients with a multidisciplinary team (MDT) diagnosis of ASyS-associated ILD, referred to each site between January 2007 and October 2017, were included. Participants were identified retrospectively from existing hospital ILD databases and medical records.
In all cases, a clinical diagnosis of ASyS had been confirmed by anti-ASyS auto-antibody positivity with associated ILD and agreed by MDT consensus (including specialist CTD rheumatologist, specialist ILD respiratory physician and ILD thoracic radiologist).
Consecutive patients presenting with IPF during the same time period were used as a comparator cohort. ASyS is an important differential diagnosis for patients presenting with idiopathic interstitial pneumonia (IIP). IPF was chosen as a comparison group for ASyS-ILD, as an archetypal IIP. All original diagnoses of IPF had been made by MDT consensus in accordance with 2011 ATS/ERS/JRS/ALAT guidelines.7 Throughout the process of data collection, the diagnosis of IPF was verified according to 2018 diagnostic criteria.8
Autoantibody testing
Anti-nuclear antibody (ANA) and anti-cytoplasmic antibody screening were performed by HEp-2 immunofluorescence (IIF). The Euroline Autoimmune Inflammatory Myopathies 16 Ag kit (Euroimmun, Luebeck, Germany) was used in all centres for cases of non-anti-Jo-1 ASyS, and for confirmation of anti-Jo1 ASyS cases originally determined by commercially available ENA methods.
Lung function testing
Pulmonary function tests were performed in accordance with ATS/ERS guidelines.9 Forced expiratory volume during first second of expiration (FEV1), forced vital capacity (FVC) and transfer factor for carbon monoxide (TLCO) were recorded at baseline and at approximately 1 year.
Outcome measures
Data were collated on patient demographics, presenting symptoms and treatment regimes, alongside lung physiology at baseline and around 1 year. High-resolution CT (HRCT) chest scans were reported by thoracic radiologists at the local centres and the predominant ILD pattern described according to the 2002 American Thoracic Society/European Respiratory Society classification criteria.1 The extent of ILD for patients with ASyS was arbitrarily quantified as <20% or ≥20% by either an ILD respiratory physician, rheumatologist or thoracic radiologist at each centre (all sections from the arch of the aorta to the top of the hemidiaphragm were evaluated). Other clinical features of ASyS were collected including mechanic’s hands, inflammatory arthritis and Raynaud’s phenomenon. Proximal myopathy and raised creatine kinase >250 were used as indicators of an active myositis.
The baseline demographics of patients with IPF were compared with those with ASyS. We also compared the clinical characteristics of patients with ASyS presenting to respiratory services with those initially presenting to rheumatology services.
Disease progression and/or treatment response for patients with ASyS at around 1 year was defined on lung function as a decline or improvement in FVC by more or less than 10% predicted compared with baseline and/or decline or improvement in TLCO by more or less than 15% predicted, as used in systemic sclerosis.10 Radiographic evolution was defined by a change in the predominant pattern of ILD on follow-up HRCT compared with baseline.
Statistical analysis
Categorical variables were presented as counts with percentages. All continuous data were non-parametric and therefore presented with medians and IQR. Differences between patient groups were evaluated using Mann-Whitney U for continuous data and Fisher’s exact test or χ2 testing for categorical data. For survival and follow-up analysis, 20 August 2019 was used as the censoring date. For all tests, a p<0.05 was considered statistically significant. Data were analysed using Prism V.8.0 (Graphpad Software, San Diego, USA).
Results
Comparison of IPF and patients with ASyS
Baseline characteristics
A total of 76 patients with ASyS-ILD (women 57%) were identified across the three sites. Patients with ASyS were younger (57 vs 77 years, p<0.001) with a female predominance (57% vs 33%, p=0.006) compared with IPF. With regards to smoking status, patients with ASyS were statistically more likely to be never smokers compared with ex/current smokers for patients with IPF (table 1).
Baseline demographics of patients with anti-synthetase syndrome (ASyS) and idiopathic pulmonary fibrosis (IPF)
Lung function parameters
Baseline FEV1 (69% vs 90%, p<0.0001) and FVC (73% vs 87%, p=0.0002) % predicted values were both significantly lower for ASyS compared with IPF, although actual FVC and FEV1 values did not differ. Baseline TLCO % predicted did not differ between the two groups (table 1).
Autoantibody results
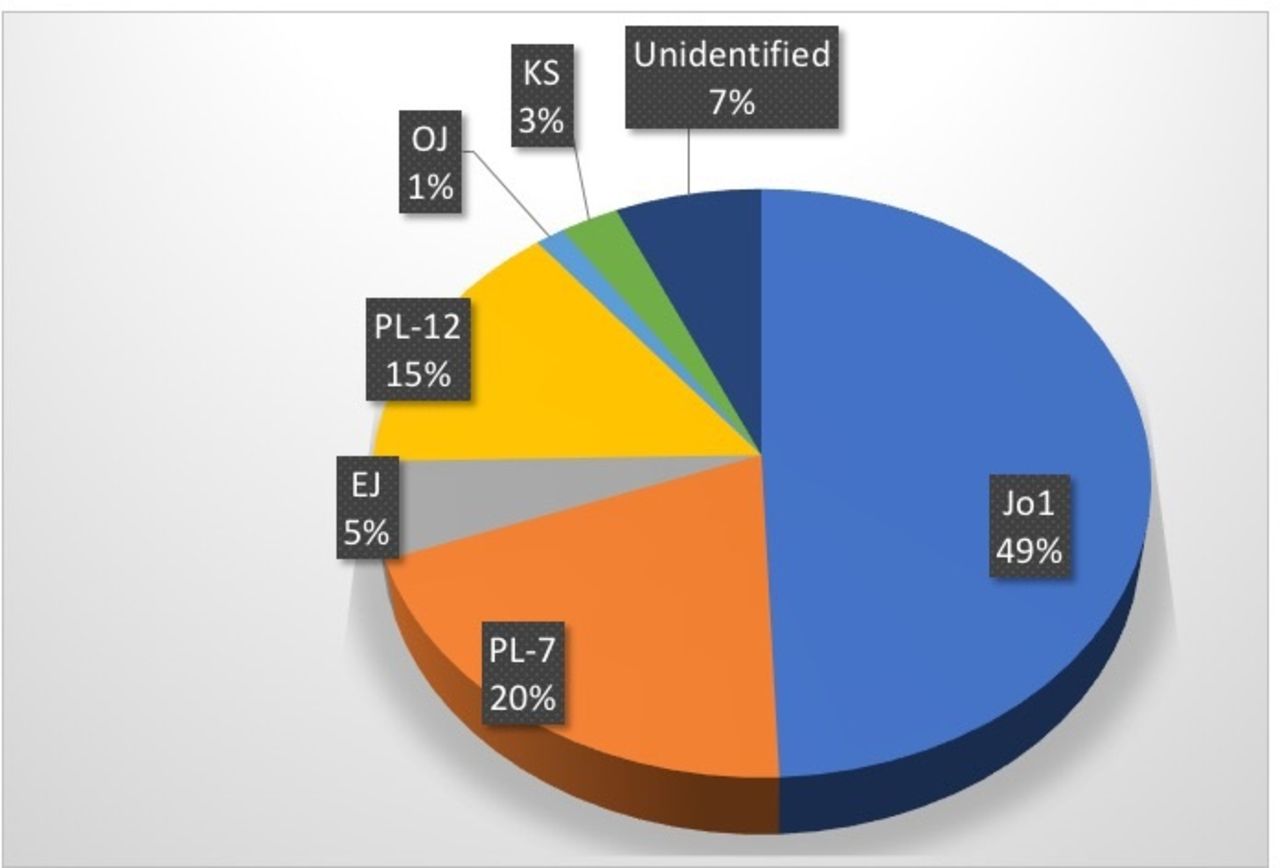
Non-specific ANA positivity was more common in IPF (39% vs 23%, p=0.038), whereas negative ANA stain but cytoplasmic pattern on indirect IIF was reported more commonly in ASyS (71% vs 0%, p<0.0001, table 1). Anti-Jo1 autoantibodies were most commonly identified in ASyS-associated ILD (49%, n=37), followed by PL-7 (20%, n=15) and PL-12 (16%, n=12). Antibodies targeting OJ, EJ and KS antibodies were identified in a minority of cases (n=7, 9%). In five patients (7%), a specific antibody could not be identified by the available laboratory techniques, despite clinical features of ASyS and cytoplasmic staining on IIF; in four of five of these cases, Ro-52 was also identified (known to coexpress with antisynthetase antibodies and are thus likely to be anti-Zo or Ha; not available for testing on this specific commercially available immunoblot) (figure 1).

Auto-antibody expression in anti-synthetase syndrome (ASys) associated interstitial lung disease (ILD). Anti-Jo-1 autoantibodies were most commonly identified in ASyS-associated ILD (49%, n=37), followed by PL-7 (20%, n=15) and PL-12 (16%, n=12). Antibodies targeting OJ, EJ and KS antibodies were identified in a minority of cases (n=7, 9%). In five patients (7%) a specific antibody could not be identified by the available laboratory techniques, despite clinical features of ASyS and cytoplasmic staining on immunofluorescence.
Radiological pattern
The majority of patients (60%) with IPF presented with a definite pattern of usual interstitial pneumonia (UIP) on HRCT. The most common radiological pattern of ILD associated with ASyS was non-specific interstitial pneumonia (NSIP) (fibrotic NSIP (fNSIP) 32%, cellular NSIP (cNSIP) 18%), although patients also presented organising pneumonia (OP) (17%), UIP (5%) or overlap radiological patterns. In this cohort, there was no significant difference in the radiological pattern of those patients with Jo-1 versus non-Jo-1-associated ILD (online supplemental table 1).
Supplemental material
Comparison of patients with ASyS presenting to respiratory versus rheumatology services
Approximately 30% patients initially presented to rheumatology services, despite the subsequent identification of ILD and with at least moderate restriction in lung physiology tests (median FVC 76% predicted (IQR 67–89), TLCO 49% predicted (IQR 39–69)). Indeed, there were no significant differences in the degree of lung function impairment at presentation in patients with ASyS-ILD presenting to rheumatology compared with respiratory services (online supplemental table 2).
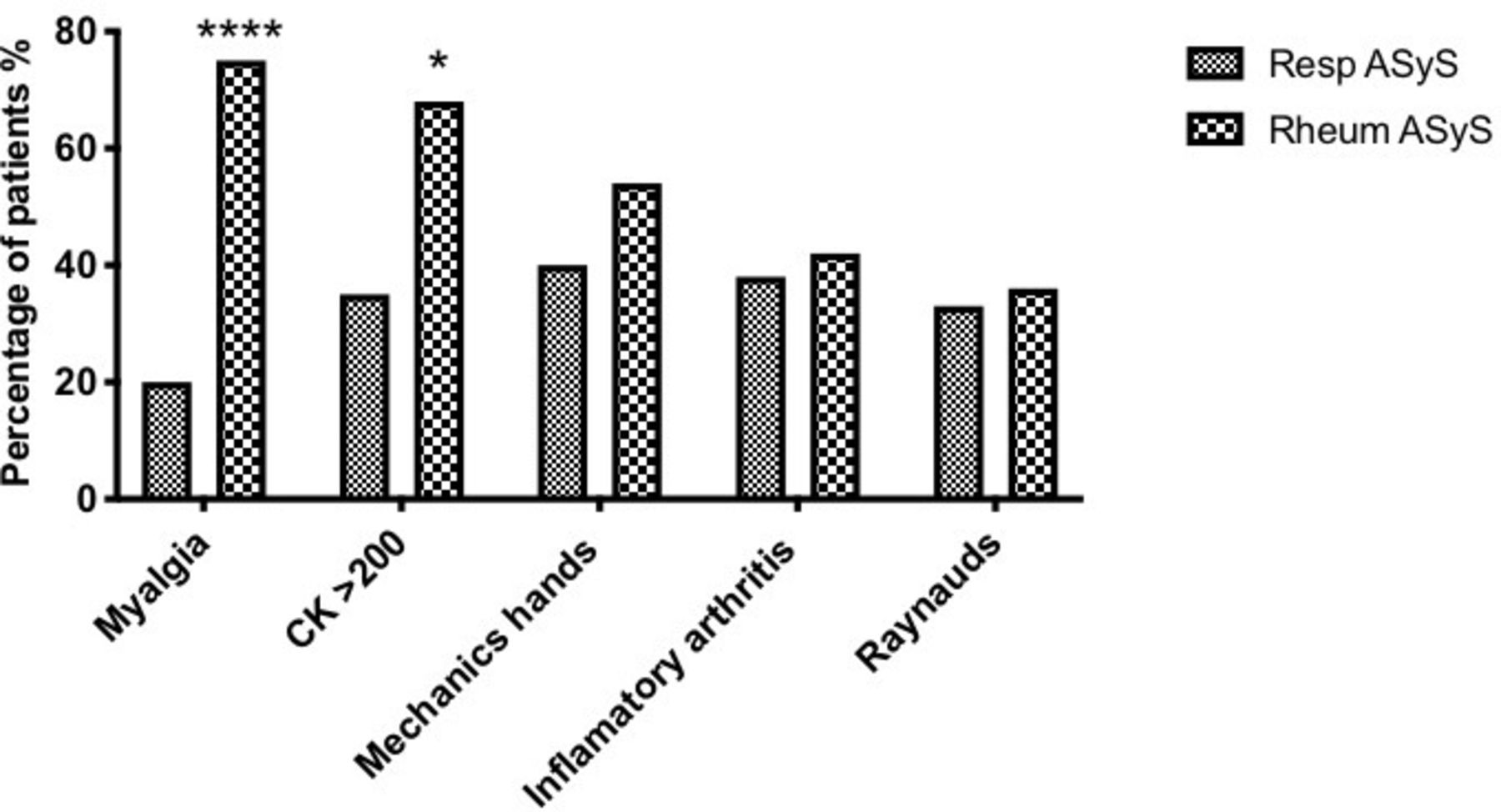
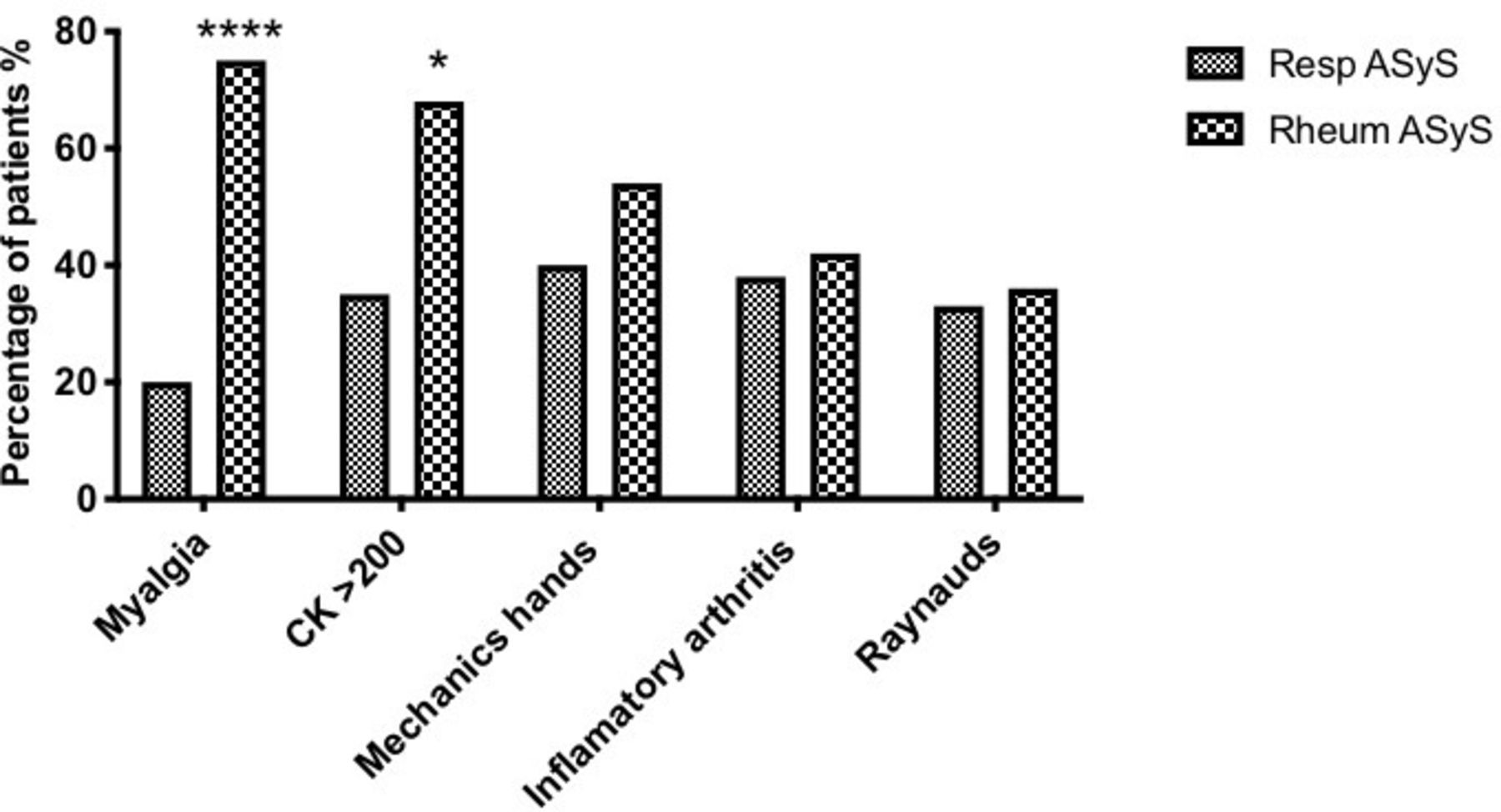
Patients with ASyS presenting initially to respiratory services were more likely to express non-Jo-1 tRNA synthetase autoantibodies (online supplemental table 3) and were less likely to describe myalgia (20% vs 75% p<0.0001) or have biochemical evidence of myositis (35% vs 68%, p<0.05), compared with those presenting to rheumatology services (p<0.05) (figure 2). The frequency of mechanic’s hands, inflammatory arthritis or Raynaud’s phenomenon did not differ between groups in terms of initial specialty review (figure 2). Other ASyS and CTD-related features were not robustly documented and could not be reliably ascertained in this retrospective data collection. Nonetheless, features such as skin rashes (n=4), Gottron’s papules (n=2), nailfold infarcts/dilation (n=3), oesophageal reflux (n=3) and fever (n=5) were additional clinical signs/symptoms recorded and attributed to ASyS in some patients but, possibly owing to the low numbers, the frequencies did not differ between groups.
{kind=link}
{kind=link}
Symptoms/signs at presentation for patients with anti-synthetase syndrome (ASyS), presenting to respiratory and rheumatology services. Patients presenting to respiratory services were less likely to describe myalgia (20% vs 75% p<0.0001****), or have biochemical evidence of myositis (35% vs 68%, p<0.05*).
Subsequent disease course in ASyS
Patients were followed up for a median of 3.97 years (IQR 2.56–5.77). Five-year survival was excellent (97%) for those patients with sufficient follow-up data available (n=30) (table 2).
Outcomes of patients with antisynthetase associated ILD (ASyS)
Detailed information of the treatments received during follow-up were available in 65 patients. The vast majority of patients (92%, n=60) received at least one form of immunomodulatory therapy during follow-up, with a median of two different medications (IQR 2–3, range 0–5) (table 2). There was no significant difference in the number of treatments used in those with Jo-1 versus non-Jo-1 ASyS-ILD (data not shown). Mycophenolate mofetil (MMF) was the most commonly used immunomodulatory treatment, with 73% of all patients (n=44/60 patients) using MMF at some point during follow-up. Cyclophosphamide induction (protocol of 6 pulses intravenously used as standard approach across the three sites) was given to 18 patients, all but one presenting with extensive lung involvement of HRCT (>20% ILD on HRCT); one patient had limited ILD (<20% extent) but rapidly worsening lung function testing. Four further patients received intravenous cyclophosphamide during their course of treatment, due to intolerance to other medications (n=1), coexistent myositis despite limited ILD (n=1) or for progressive ILD despite alternative immunomodulatory treatments (n=2). Rituximab was used in six patients with progressive disease refractory to treatment (including cyclophosphamide).
In those patients with lung function testing at 1 year following presentation, 60% (n=39/65) had a stable FVC% predicted; 31% (20/65) demonstrated a significant improvement and 9% (6/65) a significant decline. Similarly, 73% (38/52) of patients with follow-up TLCO % predicted at 1 year demonstrated stability, with 19% demonstrating a significant improvement in TLCO and 8% a significant decline (table 2). There was no significant difference in the outcome of 1-year lung function testing between those with Jo1 versus non-Jo1 antibody status (online supplemental table 4).
Interval imaging was available in 37 patients, over a median interval of 1 year (IQR 1–3.2 years). Of these, just over a third (n=14) demonstrated radiological progression, approximately one-third (n=12) showed improvement in disease extent, while there was disease stability in the remaining one-third (n=11) of patients (table 2). The size of this cohort precluded any statistical analysis based on antibody subtype.
In four patients (36%) in whom there was progression of disease, the pattern of ILD also evolved during the follow-up period (median follow-up 5.5 years (IQR 2.5–7.8)); fNSIP to UIP in n=2 patients, fNSIP to fNSIP/UIP pattern in n=1 and cNSIP/OP to UIP/OP in n=2. There was also evolution in the pattern of ILD in a minority of patients (n=3, 30%) that demonstrated radiological improvement during follow-up (median 2 years (IQR 1–3); cNSIP/OP to cNSIP n=1, cNSIP to fNSIP n=1 and OP to fNSIP/OP n=1).
Conclusion
The therapeutic management of CTD-associated ILD and idiopathic forms differs widely, thus accurate diagnosis is essential. We sought to evaluate the use of comprehensive autoantibody testing to help differentiate ASyS-associated ILD from idiopathic forms. Our findings emphasise the important concept that a negative ANA screen does not necessarily indicate autoantibody negativity and is in keeping with the findings of others.11 As expected, we identified additional patient demographics (age at presentation and gender), clinical features (myositis, mechanics hands) and radiographic features (non-UIP pattern on HRCT) that helped to differentiate ASyS from an patient with IPF cohort collected during the same period. Importantly, a proportion of patients with ASyS exists that overlap phenotypically with the classical presentation for IPF in terms of HRCT pattern, gender and age.
Our data suggest that only 5% of patients with ASyS present with an UIP pattern of fibrosis. We propose that a detailed CTD history and careful examination for potentially subtle clinical features of CTD for all patients presenting with an ILD of unknown cause but routine comprehensive CTD-specific serological screening in all patients may not be justified. We would suggest an ANA IIF (encouraging your local laboratory to report cytoplasmic staining, if present, in ANA-negative samples) as the initial serological test for those patients in which a CTD may be clinically suspected or those with non-UIP patterns of fibrosis on cross-sectional imaging; together with multidisciplinary evaluation. A cytoplasmic stain on IIF may indicate the presence of ASyS and could support efficient use of solid-phase immunoassays at reference laboratories capable of identifying non-Jo-1 ASyS autoantibodies.
The limited available current literature conflicts as to whether antibody specificity in ASyS influences phenotypes and outcomes.2 4–6 12 Hervier et al13 identified two different clusters of patients with ASyS; those with multiorgan involvement, commonly seen in anti-Jo1 disease and those with lung-limited disease, commonly seen in non-Jo-1 ASyS such as in PL-7 and PL-12 disease. In contrast, other groups have suggested a relative homogeneity in clinical features among the ASyS auto-antibody spectrum, but differences in the time of onset of these manifestations of disease (such as ILD and myositis), among patients with different subsets of ASyS autoantibodies.14 The most recent large multicentre retrospective study of more than 800 patients with ASyS, from across 10 countries, concluded that the clinical presentation and disease course of patients with ASyS were broadly similar regardless of the specific antibody present,2 although patterns of ILD were not characterised. Our findings support the findings of others in that there were no significant differences in the outcomes of patients with Jo-1 and non-Jo-1 ASyS-ILD, furthermore, the radiological pattern of patients presenting with Jo-1 or non-Jo-1 disease did not differ; NSIP being the most common presenting pattern. Patients presenting to respiratory were, however, more likely to have an amyopathic phenotype and absence of biochemical myositis compared with those presenting to rheumatology services.
In our study, approximately one-third of patients demonstrated radiological progression during follow-up, with just over a third of these patients showing an evolution in their radiological pattern towards a UIP pattern of fibrosis. Respiratory physicians therefore need a high index of suspicion to diagnose ‘lung dominant ASyS’ in patients presenting with pulmonary fibrosis of unknown cause, as they may present with only subtle clinical signs and a radiological pattern that may be radiographically indistinguishable from IPF.
There is no established treatment strategy for patients with ASyS-associated ILD; the rarity of ASyS itself, representing one of the most significant challenges for establishing evidence-based guidelines. While patients with IPF typically demonstrate inexorable decline in their lung function; our data suggest positive effects of immunosuppression with only a minority of patients having evidence of radiological progression or decline in lung function at 1 year of follow-up.
We recognise the important limitations of this study including those related to the retrospective nature of the work and the potential for missing data, although to date few prospective studies of this ASyS exist,15 presumed in part to the rarity of this condition.
Furthermore, while classification criteria for ASyS have been proposed,16 17 there may have been heterogeneity in terms of diagnostic criteria, although only cases in which an MDT consensus diagnosis was achieved were included in the analysis. As our analysis focused on ILD, we did not include patients with ASyS without ILD but the expectation is that this would have further highlighted differences in presentation in terms of clinical phenotype and secondary care referral practice.
In summary, we present a large UK multicentre cohort of patients with ASyS–ILD . Our findings highlight the rarity of myositis in patients presenting initially to respiratory services, the predominance of an NSIP pattern of fibrosis and reassuring 5-year survival. We strongly advocate the use of CTD-specific serological testing in the early assessment of patients presenting with ILD of unknown cause, alongside multidisciplinary evaluation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SLB conceptualised the paper. BM, HI, CC, HHA collected the data. HIA, LS, HG, JDP and SLB contributed their clinical cases and have been involved in drafting and revising the manuscript for content. All authors read and approved the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests JDP has received speaker’s honoraria and research grant support (>US$10 000) from Actelion Pharmaceuticals. JDP has undertaken consultancy work for Actelion Pharmaceuticals, Sojournix Pharma and Boehringer Ingelheim; outside of the submitted work. SLB received speaker’s honoraria from Boehringer Ingelheim, outside the submitted work.
Patient consent for publication Not required.
Ethics approval The study was approved by the Health Research Authority and health and Care Research Wales (HCRW), United Kingdom (IRAS 234299) and local R&D approval obtained at each site.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available. Data available upon reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.